Abstract
Objectives. To investigate the mechanisms of losartan- and exercise training-induced improvements on endothelial dysfunction in heart failure. Design. Sprague-Dawley rats subjected to left coronary artery ligation inducing myocardial infarction and heart failure were randomized to losartan treatment, high-intensity exercise training, or both. Results. Losartan, but not exercise training, reduced the heart failure-associated elevation in left ventricular end-diastolic pressure (26 ± 2 mmHg vs. 19 ± 1 mmHg after losartan). In contrast, both exercise training and losartan improved exercise capacity, by 40% and 20%, respectively; no additional effects were observed when exercise training and losartan were combined. Aortic segments were mounted on a force transducer to determine vasorelaxation. Heart failure impaired endothelium-dependent vasorelaxation, observed as a 1.9-fold reduced response to acetylcholine (EC50). Exercise and losartan improved acetylcholine-mediated vasorelaxation to the same extent, but by different mechanisms. Exercise training upregulated the nitric oxide pathway, whereas losartan upregulated a non-nitric oxide or -prostacyclin pathway; possibly involving the endothelium-dependent hyperpolarizing factor. Conclusions. Both losartan and exercise training reversed endothelial dysfunction in heart failure; exercise training via nitric oxide-dependent vasorelaxation, and losartan via an unknown mechanism that may involve endothelium-dependent hyperpolarizing factor. Thus, the combined treatment activated an additional nitric oxide- independent mechanism that contributed to reduce endothelial dysfunction.
Introduction
Endothelial dysfunction contributes to heart failure (HF) pathogenesis by increasing cardiac afterload and reducing muscle perfusion with subsequently reduced exercise capacity (Citation1,Citation2). It is characterized by a reduced capacity of the blood vessels to dilate and is caused by reduced availability of substances causing vascular smooth muscle relaxation. The main smooth muscle relaxant is endothelium-derived nitric oxide (NO), of which generation may be limited by substrate for production (L-arginine), limitation and deactivation of the catalyzer (endothelial NO synthase, eNOS) and scavenging of NO (oxidant status). However, independent of NO, also insufficient availability of endothelial-derived hyperpolarizing factors (EDHF) can contribute to decreased smooth muscle relaxation (Citation7), although the exact cellular mechanism by which this occurs remains unknown.
As such, restoration of endothelial function remains important for the management of HF. Both physical exercise and the angiotensin II type 1 receptor blocker (ARB) losartan are known to beneficially alter endothelial and overall health (Citation4,Citation5). Exercise training counteracts endothelial dysfunction by activating NO production by eNOS (Citation1), and by improving work capacity and quality of life in HF patients (Citation2,Citation5–7), whereas losartan also favorably affects the outcome of HF (Citation8,Citation9), at least partly by enhancing endothelial-dependent vasorelaxation (Citation10). Thus, both exercise training and losartan treatments may be advantageous for vascular and endothelial health in HF. However, the effect of a combination of exercise training and losartan in HF relative to treatments with either option alone remains poorly understood.
The aim of this study was therefore to determine the effect of exercise training and losartan, either each alone or in combination, on the endothelial dysfunction in HF after myocardial infarction. Moreover, we also investigated the mechanisms by which endothelial dysfunction was corrected. Our hypothesis was that both exercise training and losartan would contribute toward correcting endothelial function in HF, but by different endothelium-dependent mechanisms.
Materials and methods
Study design
Left coronary artery ligation leading to myocardial infarction and subsequent HF, or sham operation, as previously described (Citation11,Citation12), was performed in female Sprague-Dawley rats (Møllegaards Breeding Center Ltd, Denmark) during 1% isoflurane anesthesia in 70% O2/30% N2O. Buprenorphine (0.05 mg Temgesic, Reckitt and Coleman, Hull, UK) was given subcutaneously immediately and 10 hours after surgery. 7 days post-surgery, 2-dimensional short-axis echocardiography confirmed myocardial infarction. Rats were randomized to 6 different groups with 8 in each group; see . Losartan (2 g∙L− 1 ad libitum, Merck & Co., Whitehouse Station, NJ) or placebo in the drinking water was initiated 1 week after the surgery, whereas exercise training was initiated 4 weeks after the surgery, to mimic a clinically relevant treatment strategy; controls remained sedentary. 48 hours after the last exercise session, subcutaneous anesthesia was given (in mL: 0.33 haloperidol, 0.5 fentanyl, 0.5 midazolam, 0.3 ketamine hydrochloride, 0.5 water) and a pressure microtip catheter (2-Fr size Millar, Millar Instruments, Houston, TX) was inserted into the right carotid artery and advanced to the heart, whereby left ventricular (LV) end-diastolic and peak systolic pressures were measured, before euthanasia. The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996), and was approved by the Institutional Review Board.
Table I. Experimental interventions.
Maximal oxygen uptake and exercise training
After a 20-minute warm-up by running on a 25° inclined treadmill at ˜50% of the maximal oxygen uptake (VO2max), the treadmill velocity was increased progressively every 2 minutes until exhaustion. Oxygen uptake was measured continuously in a custom-made metabolic chamber, and the highest recorded value was taken as a representation of VO2max.
The endurance exercise training was performed as previously described (Citation11,Citation12). After 10-minutes of warm-up, rats were running uphill (25°) on a treadmill for 1.5 hours, alternating between 8 minutes at an exercise intensity corresponding to 85–90% of VO2max, and 2 minutes active recovery at 50–60%. Exercise was performed 5 days/week for 8 weeks, and VO2max was measured every week in the exercising animals to adjust running speed in order to maintain high intensity throughout the exercise period, whereas VO2max in sedentary rats was measured before and after the experimental period.
Vascular function
The abdominal aorta was carefully dissected with care taken to not touch the endothelium. 6 2–3 mm long segments from each rat were connected to force transducers and immersed in a 10 mL organ bath containing Krebs buffer, whereupon tension was increased stepwise to 1 gram over 75 minutes. A modified high K+ (60 mM) Krebs buffer was added and washed out again, and tension was again stabilized to 1 gram, whereby a dose of phenylephrine (3∙10 − 4 M) was added before addition of acetylcholine (10 − 4 M) to ensure reactivity. After washing and stabilizing the segments once more, the following protocols A-F were initiated; see . Briefly described, aorta segments were contracted with phenylephrine and relaxed with accumulating doses of either acetylcholine or Na+ nitroprusside (SNP), in the presence of superoxide dismutase (SOD) and/or indomethacin to inhibit superoxide and prostacyclin productions, respectively. After re-stabilizing and incubating the segments as described above, a second set of contractions and relaxations were initiated, but this time with the addition of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) or NG- nitro-L-arginine (LNNA) for 45 minutes to inhibit soluble guanylyl cyclase (sGC) and eNOS, respectively (). The chemicals were purchased from Sigma-Aldrich, except ODQ (Cayman Chemical, Ann Arbor, MI). Dose–response curves and EC50–values (the agonist concentration that evokes 50% of maximal dilatation) were obtained and areas under curves were calculated using the trapezoidal rule in Sigmaplot version 8.02. Although the abdominal aorta is a capacitor, and has little importance in the regulation of blood pressure, previous studies in our group have shown equal exercise-induced endothelial responses in the carotid artery as in the aorta (Citation13).
Statistical analysis
Mann–Whitney U test determined differences in EC50–values, and a univariate repeated-measures general linear model with the Scheffé post hoc test assessed differences in the relaxation responses. To identify changes in VO2max, the Friedman test for within-group comparisons over time and the Kruskal-Wallis test for between-group comparisons were used. p < 0.05 was considered as significant. Data are presented as mean± SEM.
Results
HF
In order to confirm development of HF after myocardial infarction due to left coronary artery ligation, we measured LV end-diastolic and peak-systolic pressures (LVEDP and LVPSP, respectively). We observed elevated LVEDP and reduced LVPSP in all post-myocardial infarction rats included in the study, and thus the presence of HF was confirmed (). Exercise training did not alter the LVEDP, although a small but insignificant reduction was noted. In contrast, losartan decreased LVEDP by 8 mmHg. None of the treatments corrected for abnormal LVPSP.
Table II. Left ventricular pressure recordings and infarct sizes.
VO2max
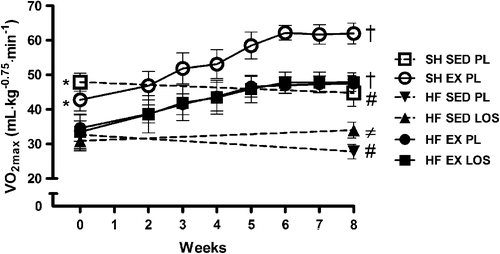
HF was associated with a ˜40% reduction in exercise capacity (VO2max) as compared to healthy sham-operated rats (). However, exercise training restored VO2max to levels comparable to sedentary sham-operated rats. The magnitude of the exercise effect was similar to that of healthy rats. Losartan alone also improved VO2max, but considerably less than exercise training (˜20%), whereas the combination of losartan and exercise training did not result in any effect beyond that of exercise training alone. This suggests that exercise training may take out the potential for improvement, such that additive strategies for increasing VO2max have no further effects. Both sham-operated and HF rats that remained sedentary and did not receive losartan decreased VO2max over the course of the study.
Figure 1. Maximal oxygen uptake (VO2max) as a measure of exercise capacity. VO2max in sham-operated (SH) rats was 40% higher (*p < 0.01) than in those with heart failure (HF) before the exercise training program (pre-test). Post-tests indicated that VO2max increased 40% by exercise training (EX) in SH and HF rats; † (p < 0.01), and by 20% in losartan (LOS)-treated sedentary (SED) HF rats compared to placebo (PL)-controlled HF rats; ≠ (p < 0.01), respectively. Note that HF EX PL and HF EX LOS curves sit on top of each other. Also, SH SED PL and HF SED PL rats decreased VO2max from pre-to post-tests; # (p < 0.05).
Acetylcholine-dependent vasorelaxation
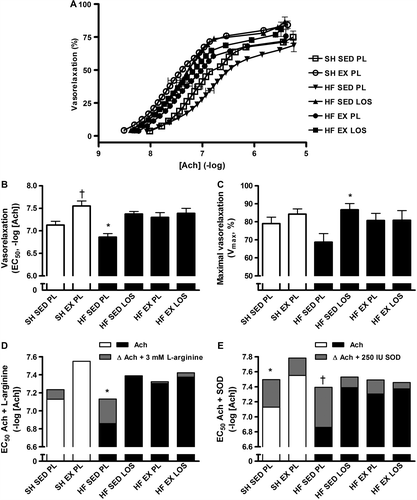
Endothelium-dependent vasorelaxation in the aorta was reduced in HF; indicating endothelial dysfunction, as evidenced by the 1.9-fold higher EC50, i.e. reduced sensitivity, in response to acetylcholine in HF compared to sham-operated rats ( and B). Exercise training, losartan, and the combination of losartan and exercise training improved vasorelaxation 2.8-fold, 3.3-fold, and 3.4-fold, respectively (p < 0.05 for all); thus, the combination of exercise training and losartan did not change the magnitude of improvement compared to either intervention alone. Furthermore, aorta segments of HF rats on losartan or exercise training also showed a trend toward higher sensitivity to acetylcholine than those of sedentary healthy rats, albeit it did not reach statistical significance. Exercise training in healthy rats also improved the acetylcholine sensitivity (EC50) and hence endothelial function. Maximal dilatation (Vmax) of the aorta tended to be reduced in HF (˜18%, p < 0.1), but restored by treatment with both losartan and exercise training ().
Figure 2. A: Heart failure (HF) reduced the acetylcholine (Ach)-mediated vasorelaxation of aorta as indicated by the right shifted dose-response curve of sedentary (SED) HF rats on placebo (PL), whereas exercise training and losartan corrected for this dysfunction. B: Sensitivity to acetylcholine (EC50); note the y-axis has a logarithmic scale. Aorta segments of the SED HF rats relaxed less than all the other groups (*p < 0.05). Interventions with exercise training (EX), losartan (LOS) or a combination of both (EX LOS) improved endothelial sensitivity to Ach to levels comparable with SED sham-operated (SH) rats, whereas SH EX rats exceeded SED SH rats (†p < 0.01). C: Maximal vasorelaxation was increased in LOS-treated SED HF rats compared to PL-controlled SED HF rats (*p < 0.05). D: EC50-values of Ach-mediated vasorelaxation (white and black bars), with delta EC50-values (grey bars) indicating difference in Ach-mediated vasorelaxation after addition of 3 mM L-arginine; this maneuver increased the response in HF-SED-PL (*p < 0.05). E: EC50-values of Ach-mediated vasorelaxation (white and black bars), with delta EC50-values (grey bars) indicating difference in Ach-mediated vasorelaxation after addition of 250 IU superoxide dismutase (SOD); this maneuver increased the responses in SH-SED-PL and HF-SED-PL (*p < 0.05 and †p < 0.01).
Acetylcholine-mediated vasorelaxation after addition of L-arginine and SOD
Addition of 3 mM L-arginine improved vasorelaxation in untreated, sedentary HF rats, observed as a 1.9-fold increase in acetylcholine sensitivity (). L-arginine-dependent effects were not seen in the other groups, although EC50 tended to improve in sham-operated sedentary rats, but this did not reach statistical significance (p = 0.06).
Incubation with 250 IU SOD increased sensitivity to acetylcholine in untreated, sedentary HF and sham-operated rats by 3.4-fold and 2.3-fold, respectively (). No significant changes occurred in the remaining groups, although a trend for improved endothelial function was observed in all of them.
Addition of L-arginine or SOD did not affect Vmax in any of the groups (data not shown). Nonetheless, as shown in and E, HF presents with a potential for improving acetylcholine-mediated vasorelaxation by interventions to increase L-arginine or to inhibit superoxides, which react with NO to form peroxynitrite. This potential is no longer observed in HF after treatment with exercise training or losartan.
Acetylcholine-mediated vasorelaxation after inhibition of prostacyclin and NO productions
Incubation with 10 − 5 M indomethacin revealed no further differences in vasorelaxation between the groups (), suggesting that prostacyclin did not contribute to the observed vasorelaxation effects presented in . In contrast, inhibition of NO-synthesis by 10 − 4 M LNNA abolished the exercise-induced increase in vasorelaxation, whereas losartan-treated rats remained with a trend (p = 0.12) toward greater vasorelaxation (). Moreover, blocking sGC with 10 − 5 M ODQ before adding accumulating doses of acetylcholine abolished vasorelaxation and instead led to a minor, but uniform contraction of 2–4% ().
Figure 3. A: Acetylcholine (Ach)-mediated vasorelaxation in the presence of 10 − 5 M indomethacin (Indo). B: Ach-mediated vasorelaxation in the presence of 10 − 4 M LNNA and 10 − 5 M Indo, with a trend (p = 0.12) toward an increase in vasorelaxation in the heart failure (HF) rats on losartan (LOS) vs. placebo (PL)-controlled sedentary (SED) or exercise training (EX) HF or sham-operated (SH) rats. C: Ach-mediated vasorelaxation in the presence of 10 − 5 M ODQ and 10 − 5 M Indo. D: Vasorelaxation due to nitric oxide (NO) was calculated by subtracting the area under curve (AUC) of the graphs in panels B and C from the AUC of panel A (A-(B + C)). HF-EX-LOS vs. HF-SED-PL and HF-SED-LOS: *p < 0.05; SH-EX-PL and HF-EX-PL vs. HF-SED-LOS, HF-SED-PL and SH-SED-PL: †p < 0.01.
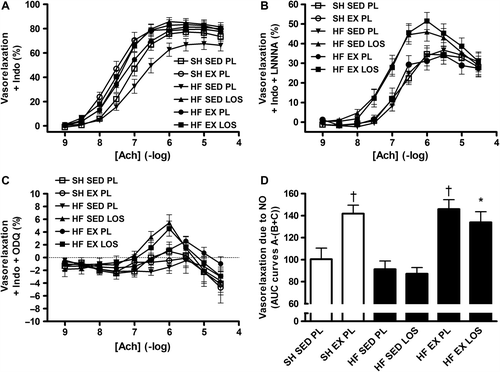
Subtracting the area under these two curves from the area under the curve of the non-NO-inhibited relaxation (curve 3A-(curves 3B + 3C)) revealed that the exercise-trained aorta segments relied ˜50% more on NO for relaxation, compared to the sedentary groups (). This was observed in both sham-operated and HF rats. These pharmacological maneuvers also suggest that losartan in contrast exerted its effect on endothelium-dependent vasorelaxation through a non-NO and non-prostacyclin-mediated effect, of which molecular identity remains unknown, but may involve EDHF. The reason this suggests EDHF as the mechanism is that it is the only known vasorelaxant downstream of acetylcholine and independent of NO and prostacyclin. As such, treatment with either losartan or exercise training reversed the endothelial dysfunction, but by different mechanisms, which were not additive when combined, suggesting a ceiling effect on endothelial-dependent vasorelaxation of either treatment alone.
Finally, incubation with 250 IU SOD significantly (p < 0.05) increased the LNNA/indomethacin-resistant vasorelaxation in all groups except in sedentary HF rats on losartan (data not shown), whereas adding 10 − 4 M SNP to aorta segments incubated with LNNA and ODQ led to ˜100% and ˜0% relaxation, respectively (data not shown).
Discussion
HF was associated with endothelial dysfunction in the abdominal aorta. However, the dysfunction was largely reversed by exercise training or by treatment with the ARB losartan, but by two distinct signaling pathways; exercise training through an NO-dependent effect and losartan through a non-NO- and non–prostacyclin effect; thus suggesting losartan may have activated EDHF. When both exercise training and losartan were combined, losartan did not inhibit the salutary effects of NO, and furthermore, it activated an additional pathway of vasoregulation that may become important in periods of exercise cessation, albeit the combination did not improve endothelial-dependent vasorelaxation further of either treatment alone. This may be explained by exercise training and losartan both individually inducing a near-maximal effect on acetylcholine-mediated endothelium-dependent vasorelaxation. Nonetheless, a combination of exercise training and losartan may provide a better strategy for long-term HF treatment as it allows periods of absence of exercise without compromising endothelial function, as well as preserving the independent effects of each treatment alone beyond the endothelium (Citation7–9).
Nitric oxide-dependent vasoregulation
By inhibiting eNOS-mediated NO-production by LNNA, prostacyclin-production by indomethacin, and sGC-cyclic guanosine monophosphate (cGMP)-dependent vasorelaxation by ODQ, the relative contributions of these pathways were assessed. This showed that impaired vasorelaxation in HF was due to reduced endothelial NO-availability, and that exercise training mainly improved vasorelaxation by stimulating NO-availability in both healthy and HF rats. This has also been suggested previously (Citation1,Citation6,Citation13). Exercise training in combination with losartan also improved endothelial function via an NO-dependent pathway; as shown previously (Citation14,Citation15), whereas in contrast, losartan alone did not stimulate NO-availability.
It has been suggested that eNOS-inhibition by L-arginine antagonists may only affect the stimulated and not the basal NO-production (Citation16). In line with this, addition of SOD in the presence of LNNA, an irreversible inhibitor with a slow dissociation rate (Citation17), still stimulated the vasorelaxation, except in losartan-treated sedentary HF rats. This points to an inability to completely block the basal NO-production, but also suggests that losartan upregulated an endothelial-mediated NO- and prostacyclin-independent vasodilator, for which the molecular identity remains unknown, but may be EDHF. As such, although the combination of exercise training and losartan did not provide observable additive effects, the combination strategy may be important inasmuch as previous experiments have suggested that the exercise-induced NO-dependent vasodilation varies greatly and exists only transiently after an exercise session (Citation18). Losartan may thus provide an additional pathway of vasorelaxation when the NO-mediated regulation is low.
Endothelial-mediated NO- and prostacyclin-independent vasorelaxation
The foremost candidate for endothelial-mediated NO- and prostacyclin-independent vasoregulation is EDHF, but its molecular identity remains unclear and may encompass several factors originating from the endothelium (Citation19); of which hydrogen sulfide (H2S) is a recent candidate (Citation20). In HF, the contribution of EDHF relative to NO for inducing vasorelaxation is increased, partly because NO is reduced (Citation21). This was also indicated in our study. The finding that losartan exerted an endothelium-dependent, but NO- and prostacyclin-independent effect, i.e. an effect attributable to stimulated EDHF, is similar to that observed after angiotensin-converting enzyme (ACE) inhibition (Citation22). Together, this suggests an angiotensin II action that is not confined to the vascular smooth muscle cell, but may also involve the endothelium.
Since blockade of the sGC-cGMP pathway by ODQ effectively removed both NO-dependent and endothelium-dependent but NO- and prostacyclin-independent, possibly EDHF-mediated, vasorelaxation, it opens up the possibility that NO and EDHF or its equivalent may initiate similar sGC-cGMP-dependent reactions in the smooth muscle cell. This is in line with the reports of angiotensin II negatively affecting sGC independent of NO (Citation23). Finally, we also observed that the NO- and prostacyclin-independent contribution to endothelium-mediated vasorelaxation was reduced when exercise training was introduced to HF rats on losartan, indicating that activated NO prevailed as the main vasodilator.
Conclusion
Endothelial dysfunction in HF was reversed by treatments involving exercise training and the ARB losartan, either alone or in combination. However, whereas exercise training improved endothelial function by restoring NO-production, losartan upregulated a different intra-endothelial pathway that is NO- and prostacyclin-independent. When losartan and exercise training were combined, no additional vasorelaxation occurred, but NO prevailed as the main vasodilator. Nonetheless, losartan might activate an additional mechanism for vasorelaxation that supplements NO-dependent vasorelaxation, which may be important when and if NO production is compromised.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
The authors are indebted to MSD for providing losartan and to the Norwegian Council on Cardiovascular Diseases (OJK, UW, OE), the St Olavs Hospital (OJK, UW, OE), the British Heart Foundation (OJK), the Norwegian Research Council (UW), and the K.G. Jebsen Foundation (UW) for funding.
References
- Hambrecht R, Adams V, Erbs S, Linke A, Krankel N, Shu Y, et al. Regular physical activity improves endothelial function in patients with coronary artery disease by increasing phosphorylation of endothelial nitric oxide synthase. Circulation. 2003;107:3152–8.
- Hambrecht R, Gielen S, Linke A, Fiehn E, Yu J, Walther C, et al. Effects of exercise training on left ventricular function and peripheral resistance in patients with chronic heart failure: a randomized trial. JAMA. 2000;283:3095–101.
- Fitzgerald SM, Bashari H, Cox JA, Parkington HC, Evans RG. Contributions of endothelium-derived relaxing factors to control of hindlimb blood flow in the mouse in vivo. Am J Physiol Heart Circ Physiol. 2007;293:H1072–82.
- Hambrecht R, Wolf A, Gielen S, Linke A, Hofer J, Erbs S, et al. Effect of exercise on coronary endothelial function in patients with coronary artery disease. N Engl J Med. 2000;342:454–60.
- Mann DL, Deswal A, Bozkurt B, Borre-Amione G. New therapeutics for chronic heart failure. Annu Rev Med. 2002;53:59–74.
- Lindsay DC, Jiang C, Brunotte F, Adamopoulos S, Coats AJ, Rajagopalan B, et al. Impairment of endothelium dependent responses in a rat model of chronic heart failure: effects of an exercise training protocol. Cardiovasc Res. 1992;26:694–7.
- Wisloff U, Stoylen A, Loennechen JP, Bruvold M, Rognmo M, Haram PM, et al. Superior cardiovascular effect of aerobic interval training versus moderate continuous training in heart failure patients: a randomized study. Circulation. 2007;115:3086–94.
- Pitt B, Poole-Wilson PA, Segal R, Martinez FA, Dickstein K, Camm AJ, et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: randomised trial-the Losartan Heart Failure Survival Study ELITE II. Lancet. 2000;355:1582–7.
- Pitt B, Segal R, Martinez FA, Meurers G, Cowley AJ, Thomas I, et al. Randomised trial of losartan versus captopril in patients over 65 with heart failure (Evaluation of Losartan in the Elderly Study, ELITE). Lancet. 1997;349:747–52.
- Ferrario CM. Use of angiotensin II receptor blockers in animal models of atherosclerosis. Am J Hypertens. 2002; 15:9S–13S.
- Kemi OJ, Hoydal MA, Haram PM, Garnier A, Fortin D, Ventura-Clapier R, et al. Exercise training restores aerobic capacity and energy transfer systems in heart failure treated with losartan. Cardiovasc Res. 2007;76:91–9.
- Kemi OJ, MacQuaide N, Hoydal MA, Ellingsen O, Smith GL, Wisloff U. Exercise training corrects control of spontaneous calcium waves in hearts from myocardial infarction heart failure rats. J Cell Physiol. 2012;227:20–6.
- Kemi OJ, Haram PM, Wisloff U, Ellingsen O. Aerobic fitness is associated with cardiomyocyte contractile capacity and endothelial function in exercise training and detraining. Circulation. 2004;109:2897–904.
- Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94:3341–7.
- Crozier I, Ikram H, Awan N, Cleland J, Stephen N, Dickstein K, et al. Losartan in heart failure. Hemodynamic effects and tolerability. Losartan hemodynamic study group. Circulation. 1995;91:691–7.
- Kubo SH, Rector TS, Bank AJ, Raij L, Kraemer MD, Tadros P, et al. Lack of contribution of nitric oxide to basal vasomotor tone in heart failure. Am J Cardiol. 1994;74:1133–6.
- Furfine ES, Harmon MF, Paith JE, Garvey EP. Selective inhibition of constitutive nitric oxide synthase by L-NG-nitroarginine. Biochemistry. 1993;32:8512–7.
- Haram PM, Adams V, Kemi OJ, Brubakk AO, Hambrecht R, Ellingsen O, Wisløff U. Time-course of endothelial adaptation following acute and regular exercise. Eur J Cardiovasc Prev Rehabil. 2006;13:585–91.
- Feletou M, Vanhoutte PM. Endothelium-dependent hyperpolarizations: past beliefs and present facts. Ann Med. 2007;39:495–516.
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science. 2008;322:587–90.
- Malmsjo M, Bergdahl A, Zhao XH, Sun XY, Hedner T, Edvinsson L, Erlinge D. Enhanced acetylcholine and P2Y-receptor stimulated vascular EDHF-dilatation in congestive heart failure. Cardiovasc Res. 1999;43:200–9.
- Westendorp B, Schoemaker RG, van Gilst WH, Buikema H. Improvement of EDHF by chronic ACE inhibition declines rapidly after withdrawal in rats with myocardial infarction. J Cardiovasc Pharmacol. 2005;46:766–72.
- Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res. 2002;90:E58–65.