
Abstract
Objectives: Flecainide is class Ic antiarrhythmic agent that was found to increase the risk of sudden cardiac death. Arrhythmic responses to flecainide could be precipitated by exercise, suggesting a role played by inappropriate rate adaptation of ventricular repolarization. This study therefore examined flecainide effect on adaptation of the QT interval and ventricular action potential duration (APD) to abrupt reductions of the cardiac cycle length. Design: ECG and ventricular epicardial and endocardial monophasic APD were recorded in isolated, perfused guinea-pig heart preparations upon a sustained cardiac acceleration (rapid pacing for 30 s), and following a single perturbation of the cycle length evoked by extrasystolic stimulation. Results: Sustained increase in heart rate was associated with progressive bi-exponential shortening of the QT interval and APD. Flecainide prolonged ventricular repolarization, delayed its rate adaptation, and decreased the amplitude of QT interval and APD shortening upon rapid cardiac pacing. During extrasystolic stimulation, flecainide attenuated APD shortening in premature ventricular beats, with effect being greater upon using a longer basic drive cycle length (S1-S1=550 ms versus S1-S1=300 ms). Conclusions: Flecainide-induced arrhythmia may be partly accounted for by attenuated adaptation of ventricular repolarization to sudden changes in cardiac cycle length provoked by transient tachycardia or ectopic beats.
Introduction
In human patients, a spontaneous episode of malignant ventricular tachyarrhythmia is often precipitated by sudden changes in cardiac cycle length that may be provoked by a single premature cardiac contraction or a series of rapid ectopic beats.[Citation1] Holter ECG recordings in victims of sudden arrhythmic death demonstrate that cardiac arrest is typically preceded by abrupt heart rate acceleration or oscillatory variations of the R-R interval.[Citation1,Citation2] In line with these observations, studies with programmed ventricular stimulation have shown that abrupt changes in ventricular activation rate facilitate macro-reentry within the His-Purkinje system, thus increasing inducibility of arrhythmia.[Citation3]
These clinical findings may be partly accounted for by the inadequate adaptation of ventricular repolarization to sudden changes in cardiac activation rate. In physiological conditions, an abrupt shortening of the cardiac cycle contributes to reduction in ventricular action potential duration, an effect that ensures sufficiently long diastolic interval in order to maintain appropriate ventricular filling in tachycardia. The prolongation of diastolic interval also eliminates the possibility for superposition of the next ventricular depolarization on the T wave of a preceding beat (the “R-on-T phenomenon”), which would otherwise serve as a trigger for inducing the re-entrant excitation. Consequently, attenuated shortening of ventricular repolarization upon sudden changes in activation rate may have maladaptive value, and set a stage for arrhythmia. In support of this notion, a protracted rate adaptation and longer QT interval values during exercise have been associated with a greater risk to develop exercise-induced arrhythmia in patients with coronary artery disease.[Citation4]
It is uncertain whether the rate adaptation of ventricular repolarization is affected by pharmacological treatments that increase arrhythmic susceptibility. Flecainide is class Ic antiarrhythmic agent that was found to produce serious pro-arrhythmic effects in the Cardiac Arrhythmia Suppression Trial (CAST).[Citation5] In patients with healed myocardial infarction, the risk of arrhythmic death was significantly greater in flecainide-treated group as compared to placebo controls.[Citation5] The present study was designed to test the hypothesis that arrhythmogenic responses to flecainide may be partly attributed to abnormal adaptation of ventricular repolarization to sudden variations in the cycle length. In support of this hypothesis, although flecainide was originally characterized as a potent Na+ channel blocker, it can also affect ionic currents that govern ventricular repolarization,[Citation6,Citation7] and prolong QT interval in cardiac patients.[Citation8,Citation9] Furthermore, there are clinical case reports indicating that flecainide-induced ventricular tachyarrhythmias have a propensity to occur during exercise testing;[Citation10,Citation11] this raises a possibility that drug-induced abnormalities in the rate adaptation of ventricular repolarization may play a role in creating arrhythmic substrate.
In this study, the aforementioned hypothesis was tested by exploring flecainide effects on the rate adaptation of QT interval and monophasic action potential duration in isolated, perfused guinea-pig heart preparations. The rate adaptation of ventricular repolarization was examined upon a sustained increase in cardiac activation rate by rapid pacing, and upon a single alteration in cardiac cycle length produced by extrasystolic stimulation.
Material and methods
This study complies with the European Community Guidelines for the Care and Use of Experimental Animals, and was approved by the institutional Animal Ethics Screening Committee (clearance number: 2010/561-1799). The Male Dunkin–Hartley guinea-pigs (supplied by Charles River, Sulzfeld, Germany) weighing 400–500 g were allowed to acclimate to the housing conditions, with free access to food and tap water, for at least seven days prior to entry into the study.
Isolated, Langendorff-perfused heart preparations
The experiments on isolated, perfused hearts were performed as described previously.[Citation12,Citation13] The guinea-pigs were anesthetized with sodium pentobarbital (50 mg/kg i.p.), their chests were opened, and the hearts were immediately excised and mounted on a Langendorff perfusion set-up (Hugo Sachs Elektronik-Harvard Apparatus GmbH, March-Hugstetten, Germany). The hearts were perfused via the aorta with carefully filtered, warmed physiological saline solution saturated with 95%O2 and 5%CO2. The perfusion solution contained (in mM) 118.0 NaCl; 4.7 KCl; 2.5 CaCl2; 25 NaHCO3; 1.2 KH2PO4; 1.2 MgSO4; and 10.0 glucose, and had a pH of 7.4. The coronary perfusion pressure was measured with an ISOTEC pressure transducer (Harvard Apparatus, Holliston, MA) that was connected to the aorta block of the set-up. The coronary flow-rate was determined using an ultrasonic flowmeter probe (Transonic Systems Inc., Ithaca, NY) placed just above the aortic cannula. The electrical activity of the heart preparations was assessed from the volume-conducted ECG as well as monophasic action potential (MAP) recordings. Both ECG and MAP signals were amplified, filtered at high cut-off (1 kHz) and digitized at a sampling frequency of 5 kHz. Throughout the experiments, the heart preparations were kept immersed in the temperature-controlled, perfusate-filled chamber to minimize thermal loss. Aortic pressure, coronary flow rate, ECG and ventricular MAPs were continuously monitored using a 16-channel PowerLab system (ADInstruments, Oxford, UK).
Electrophysiological recordings and electrical stimulation protocols
Drug-induced arrhythmogenic responses are facilitated upon bradycardia associated with atrioventricular (AV) block.[Citation14] Furthermore, in heart preparations with AV block, the ventricular pacing at variable intervals could be better maintained owing to the lack of competitive stimulation from the intrinsic cardiac pacemaker. Taking into account these considerations, in the present study, both atria were removed, and the AV node was crushed mechanically with forceps prior to taking electrophysiological recordings. A 30 min stabilization period was allowed prior to starting data collection.
Monophasic action potentials were recorded using two pressure contact electrodes attached to the basal epicardial surface in the left ventricular (LV) and right ventricular (RV) chambers. Endocardial MAP was recorded using a stick-shaped electrode introduced into the LV cavity via the AV orifice. The MAP duration was measured at 90% repolarization (APD90). Electrical stimulations were applied at the LV epicardial base using 2 ms rectangular pulses of twice diastolic threshold current generated by a programmable stimulator (Hugo Sachs Electronik-Harvard Apparatus GmbH, March-Hugstetten, Germany). Rate adaptation of ventricular repolarization was assessed by measuring QT interval and APD90 changes upon an abrupt alteration in cardiac cycle length that was induced by implementing two different electrical stimulation protocols.
The first protocol assessed the adaptation of repolarization to a sustained increase in cardiac activation rate. For this purpose, spontaneously beating heart preparations with a running cycle length of 603 ± 30 ms were subjected to the 30 s rapid LV pacing (a train of 100 pulses applied at S1-S1=300 ms), which produced a two-fold increase in cardiac activation rate (from ∼100 beats/min to 200 beats/min). QT interval, as well as APD90 values determined at distinct ventricular recording sites were measured in each cardiac beat during the tachypacing period, and then plotted as a function of time from the beginning of stimulation. The obtained plots of the rate adaptation of ventricular repolarization were fitted using double-exponential function: y = y0 + A1exp(-DI/τ1) + A2exp(-DI/τ2), where y represents APD90, y0 is a free-fitting variable, A1 and A2 are the amplitudes, and τ1 and τ2 are the time constants of the fast (A1 and τ1) and slow (A2 and τ2) exponential components obtained by a least squares fit. The curve fitting was performed using Igor Pro 6.0 software (WaveMetrics, Inc., Portland, OR).
The second protocol assessed the adaptation of action potential duration to a single perturbation in cardiac cycle length produced by extrasystolic beat. The protocol enrolled programmed ventricular stimulation, wherein a burst of 10 regular (S1) pulses was followed by an extrastimulus (S2) applied at variable coupling intervals, starting with S1-S2=550 ms. The S1-S2 interval was then reduced in 25 ms steps till 500 ms, then in 20 ms steps till 200 ms, and finally in 5–10 ms until the extrastimulus failed to capture the ventricle. The stimulations were repeated at two different basic drive cycle lengths (S1-S1 interval of 550 and 300 ms), and APD90 values determined in extrasystolic beats were plotted versus S1-S2 interval in order to assess dynamic changes in repolarization.
Drug infusion
For precise dosing, flecainide infusions were performed at a rate of 0.3 ml/min using a calibrated infusion pump, while perfusing the heart preparations at a constant coronary flow rate of 15 ml/min, as described previously.[Citation12,Citation14] Flecainide was infused over 30 min at a dose of 1.5 μM. Assuming the range of therapeutic concentrations of 0.5–2.5 μM, and plasma protein binding of 40%,[Citation15] the dose of flecainide used in this study is close to the maximum plasma levels of this agent determined in cardiac patients.
Data analysis
Data are expressed as means ± standard errors of the mean. Paired t-tests were used to compare samples forming two data sets, and repeated measures ANOVA was used for multiple comparisons. p values less than 0.05 were considered to be significant.
Results
Adaptation of ventricular repolarization to an abrupt increase in cardiac beating rate
shows representative ECG and monophasic action potential recordings obtained upon initiation of the rapid cardiac pacing in spontaneously beating heart preparation, and shows composite data that illustrate the rate adaptation of ventricular repolarization in 10 experiments. At baseline, the running cardiac cycle length in spontaneously beating hearts was 603 ± 30 ms, the QT interval was 203 ± 3 ms, and APD90 determined at LV epicardium, LV endocardium, and RV epicardium were 165 ± 2 ms, 173 ± 2 ms, and 174 ± 3 ms, respectively. Pacing at accelerated rate (100 pulses at S1–S1=300 ms) was associated with progressive reduction in QT interval and APD90 measured at distinct ventricular recording sites (). At the 30 s stimulation time point, the overall magnitude of shortening of the QT interval was 25 ± 2 ms, and APD90 determined at LV epicardium, LV endocardium, and RV epicardium were reduced by 19 ± 2 ms, 20 ± 2 ms, and 21 ± 2 mss, respectively.
Figure 1. Representative ECG and monophasic action potential recordings obtained upon an abrupt acceleration of cardiac beating rate. ECG, left ventricular (LV) epicardial (epi), LV endocardial (endo), and right ventricular (RV) epicardial monophasic action potentials (MAP) were recorded during spontaneous beating (the first beat in all panels), and upon initiation of rapid cardiac pacing. Spontaneous running cardiac cycle length was 610 ms, and the cycle length during pacing was 300 ms. Vertical dotted lines on ECG show the moments of regular stimulus (S1) application. The numbers (ms) below traces show beat-to-beat changes in QT interval and APD90 at epicardial and endocardial recording sites. Note the adaptation of ventricular repolarization to increased cardiac activation rate, as indicated by about 20 ms shortening in QT interval and APD90 over first seven paced beats, compared to the values determined prior to S1-S1 stimulation.
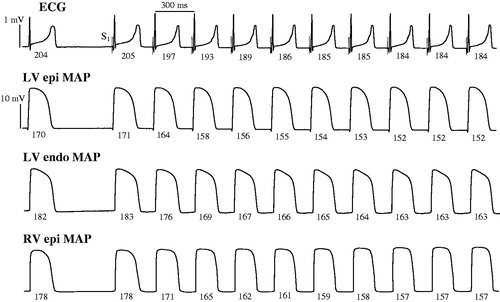
Figure 2. Rate adaptation plots of the QT interval and ventricular action potential duration determined at baseline and upon flecainide infusion. A train of 100 regular pulses at S1-S1 interval of 300 ms (total stimulation time is 30 s) was applied at LV epicardium, and QT interval (panel A), and action potential duration (APD90) determined at LV epicardium (panel B), LV endocardium (panel C), and RV epicardium (panel D) were measured in each S1 beat and plotted versus time from the beginning of stimulation. The plots obtained at baseline (squares) and upon flecainide infusion (circles) were fitted by a double-exponential function.
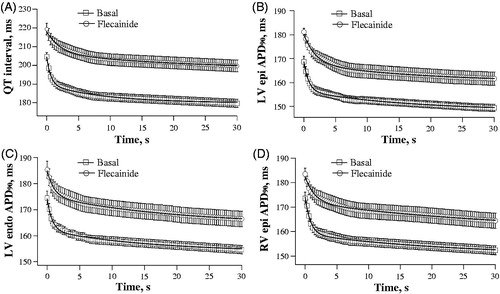
Throughout a period of rapid cardiac pacing, the duration of ventricular repolarization followed a bi-exponentially decaying time course, wherein 60–70% of the total reduction in QT interval and APD90 value was achieved during the first 5–7 cardiac cycles; this was followed by a gradual monotonic decrease in these parameters over subsequent S1–S1 intervals ( and ). The fast and slow time constants (τ) of the rate-dependent shortening of QT interval, as determined from a bi-exponential fit, were 0.62 ± 0.05 s and 12 ± 1 s. With APD90 rate adaptation, the fast time constant was close to 1 s (LV epicardium: 0.76 ± 0.06 s; LV endocardium: 0.70 ± 0.05 s; RV epicardium: 0.84 ± 0.07 s), and the slow time constant was around 15–18 s (LV epicardium: 16 ± 1 s; LV endocardium: 15 ± 1 s; RV epicardium: 18 ± 1 s).
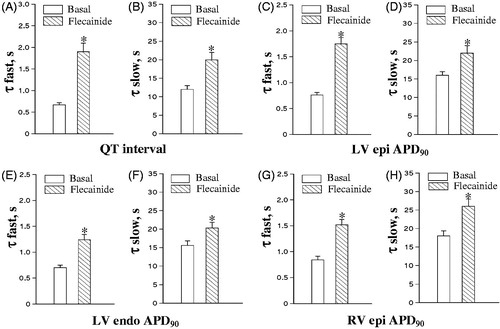
During spontaneous beating, flecainide had no effect on the running cardiac cycle length, but prolonged the QT interval (from 203 ± 3 to 220 ± 3 ms, p = 0.0003), and increased APD90 at LV epicardium (from 165 ± 2 to 180 ± 2 ms, p < 0.0001), LV endocardium (from 173 ± 2 to 186 ± 4, p = 0.01), and RV epicardium (from 174 ± 3 to 184 ± 2 ms, p = 0.007). Consequently, the QT interval and ventricular APD90 rate adaptation plots obtained upon rapid cardiac pacing were shifted upwards in flecainide-treated heart preparations relative to the basal values (). Importantly, flecainide was found to delay the adaptation of ventricular repolarization to accelerated activation rate, as evidenced by significantly increased values of the rapid and slow time constants of the QT interval shortening (, panels A and B), as well as APD90 shortening assessed at distinct ventricular recording sites (, panels C to H), upon drug infusion. Furthermore, flecainide reduced the magnitude of changes in duration of ventricular repolarization over the initial rapid phase of the rate adaptation, as evidenced by a decrease in amplitude of the fast component of the QT interval shortening (from 15 ± 1 to 11 ± 1 ms, p = 0.009), and the fast component of APD90 shortening assessed at LV epicardium (from 12 ± 1 to 8 ± 1 ms, p = 0.04), LV endocardium (from 12 ± 1 to 9 ± 1 ms, p = 0.01), and RV epicardium (from 15 ± 1 to 11 ± 1 ms, p = 0.01). Flecainide, however, had no effect on the amplitude of the slow component of the rate adaptation of QT interval and ventricular APD90 (data not shown).
Figure 3. Effects of flecainide on the kinetics of rate adaptation of ventricular repolarization. The time constants (τ) of the fast and slow component of the adaptation of QT interval and action potential duration (APD90) to increased cardiac beating rate were determined from the double-exponential fits shown in . *p < 0.05 versus corresponding basal value.
Changes in action potential duration in extrasystolic beats
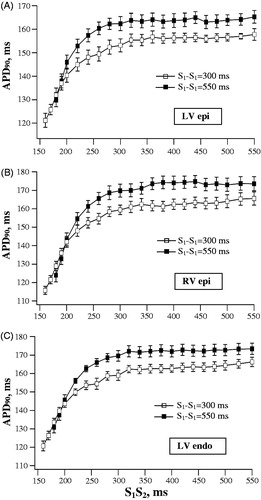
illustrates the programmed ventricular stimulation protocol that was used to assess alterations in APD90 produced by extrastimulus application at variable S1–S2 intervals, and shows the composite data from basal recordings. APD90 to S1–S2 interval relationships were examined while using two different basic drive cycle lengths (550 and 300 ms; S1–S1 interval in ). In this setting, the rate adaptation of repolarization in extrasystolic beats was manifested in exponential APD90 shortening upon progressive reduction in S1–S2 interval from 550 ms to values (160–180 ms) preceding the refractory period (). The characteristics of the rate adaptation, nevertheless, were partly determined by the basic drive cycle length. In basal conditions, APD90 in the last regular beat prior to S2 application was invariably longer at S1-S1 interval of 550 versus 300 ms, as assessed at LV epicardium (162 ± 3 versus 153 ± 2 ms), LV endocardium (171 ± 3 versus 159 ± 2 ms), and RV epicardium (171 ± 3 versus 158 ± 3 ms). Consequently, at identical coupling stimulation intervals (over a range of S1-S2 values greater than 240 ms), APD90 in extrasystolic beats was found to be longer while using the basic drive cycle length of 550 ms as compared to 300 ms (). This difference accounts for a gap in between APD90 plots obtained at S1–S1=550 ms versus S1-S1=300 ms, when the values are presented in the same coordinates ().
Figure 4. Extrasystolic stimulation protocol applied at variable basic drive cycle length. Left ventricular epicardial monophasic action potentials (MAP) were recorded upon programmed S1-S2 stimulations. Each MAP recording shows the last three regular (S1) beats in a basic drive train, followed by extrasystolic (S2) beat evoked at S1-S2 coupling stimulation intervals of 500 ms (panel A), 400 ms (panel B), and 260 ms (panel C). The moments of regular and extrasystolic stimulations are indicated by vertical dotted lines and dashed lines, respectively. The cycle length (S1-S1 interval) used in a basic drive train was 550 ms (upper fragment in each panel) and 300 ms (lower fragment in each panel). The numbers (ms) above MAP recordings indicate action potential duration at 90% repolarization (APD90) in regular and extrasystolic beats. Note that APD90 in regular beats is longer while pacing at S1-S1 cycle length of 550 ms as compared to 300 ms. Also note that at comparable S1-S2 coupling stimulation intervals, APD90 in extrasystolic beat is greater when S2 is applied after a train of pulses with S1-S1=550 mss, as compared to stimulations at S1-S1=300 ms.
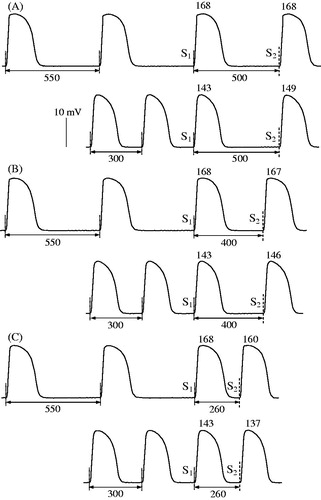
Figure 5. Basal relationships between action potential duration and S1-S2 coupling stimulation interval in extrasystolic beats. Left ventricular (LV) epicardial (epi), right ventricular (RV) epicardial, and LV endocardial (endo) APD90 were determined in extrasystolic beats evoked over a wide range of S1-S2 coupling stimulation intervals, while using the basic drive cycle length (S1-S1) of 300 ms and 550 ms.
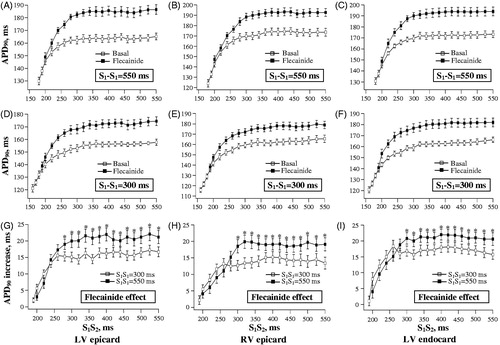
Flecainide prolonged APD90 in a train of regular beats evoked at S1-S1 interval of 550 ms (LV epicardium: from 162 ± 3 to 181 ± 3 ms, p = 0.0003; LV endocardium: from 171 ± 3 to 190 ± 3 ms, p = 0.0003; RV epicardium: from 171 ± 3 to 189 ± 3 ms, p = 0.005), and at S1-S1 interval of 300 ms (LV epicardium: from 153 ± 3 to 168 ± 3 ms, p = 0.002; LV endocardium: from 159 ± 2 to 174 ± 3 ms; p = 0.003; RV epicardium: from 158 ± 3 to 172 ± 4 ms; p = 0.01). The relative increase in APD90 by flecainide was greater at S1-S1= 550 ms versus S1-S1=300 ms (LV epicardium: 19 ± 2 versus 15 ± 1 ms, p = 0.04; LV endocardium: 19 ± 2 versus 15 ± 2 ms, p = 0.04; RV epicardium: 18 ± 1 versus 14 ± 1 ms, p = 0.01).
During extrasystolic stimulations, flecainide infusion was associated with attenuated shortening of ventricular repolarization upon S2 application. Indeed, shows that over a wide range of S1-S2 intervals, flecainide significantly increased extrasystolic APD90 values determined at distinct ventricular recording sites (, panels A to F). The rate-dependency of drug effect upon repolarization in regular beats, as outlined above, was also evident upon measuring the extrasystolic APD90 values. In S2-evoked beats, flecainide-induced lengthening in APD90 was found to be greater when using the basic drive cycle length of 550 ms as compared to S1-S1=300 ms (, panels G, H and I).
Figure 6. Effects of flecainide on action potential duration in extrasystolic beats. The data are presented as described in the legend for . Flecainide effects on APD90 in extrasystolic beats were assessed while using the basic drive cycle length of 550 ms (panels A, B and C) and 300 ms (panels D, E and F), as illustrated by the stimulation protocol shown in . The relative increase in extrasystolic APD90 value by flecainide was determined at each S1-S2 stimulation interval, and plotted separately in panels G, H and I. *p < 0.05 versus corresponding basal value (in panels G, H and I).
Discussion
The results of this study suggest that flecainide produces substantial changes in adaptation of ventricular repolarization to sudden variations in cardiac cycle length. During sustained cardiac acceleration, flecainide prolongs QT interval and action potential duration, increases the fast and slow time constant of the rate adaptation, and decreases the magnitude of QT interval and APD90 reduction over the initial rapid phase of the rate adaptation. During a single perturbation of the cycle length evoked by extrasystolic stimulation, flecainide prolonged APD90 in premature beats, thereby attenuating S2-evoked abbreviation of ventricular repolarization. Interestingly, flecainide effect on APD90 in both regular and extrasystolic beats was dependent on the basic drive cycle length, being greater at S1-S1=550 ms as compared to S1-S1=300 ms.
Previous studies reported on flecainide effects upon APD90 adaptation to changes in steady-state cardiac beating rate assessed over a wide range of S1-S1 pacing intervals (dynamic electrical restitution).[Citation22,Citation23] In contrast, the focus of the present study was on adaptation of ventricular repolarization to sudden increase in cardiac activation rate (i.e. APD90 and QT accommodation). These two methodologies are thought to uncover different aspects of the cardiac rate adaptation – the accommodation effect is determined by the time needed for APD90 and QT interval to achieve a stable shortening upon a given increase in cardiac activation rate, whereas the electrical restitution reflects how variable is the ventricular action potential duration over the whole range of stimulation frequencies that could be followed by cardiac preparation upon dynamic pacing. The assessments of APD90 changes upon an abrupt cardiac acceleration may not necessarily be concordant to those obtained with analysis of electrical restitution. For example, patients with congenital long QT syndrome typically show a protracted QT adaptation to an abrupt increase in heart rate, although the electrical restitution assessments reveal a steepened slope of QT dynamicity (i.e. a larger QT change in response to a given prolongation of cardiac cycle) compared to control subjects.[Citation4,Citation16,Citation17]
Adaptation of ventricular repolarization to abrupt shortening of the cardiac cycle is manifested in reduced action potential duration that results from enhanced outward K+ current.[Citation16] During extrasystolic stimulation, APD90 shortens immediately upon a reduction of the preceding diastolic interval, an effect ascribed to incomplete recovery of IK, the delayed rectifier K+ current, after previous regular beat.[Citation16] In contrast, sudden sustained rate acceleration is associated with more complex adaptive changes of repolarization, whereby a rapid drop in APD90 seen in the first few cardiac cycles is followed by a slow phase of monotonic APD90 shortening.[Citation16,Citation18] The latter is thought to be attributed to slowly developing alterations in ionic concentrations on both sides of the cardiac myocyte membrane, including K+ accumulation in extracellular clefts, as well as increased intracellular Na+ and Ca2+ levels.[Citation18] Collectively, these changes contribute to increased net repolarizing current in ventricular cells, which is mediated via a complex mechanism including effects on the Na+-Ca2+ exchange, Na+-K+ pump, and inactivation of the L-type Ca2+ current.[Citation16,Citation18]
Assuming a pivotal role of increased net repolarizing current in APD90 rate adaptation, flecainide effects reported in the present study may be attributed to the overall reduction in ventricular repolarization reserve. In this regard, previous works have shown that over a range of therapeutic concentrations, flecainide decreases IKr, the rapid component of the delayed rectifier K+ current, in hERG-expressing cell lines,[Citation19] and in single ventricular myocytes from guinea-pig.[Citation7] and cat [Citation6] A reduction in IKr by this agent appears to be of clinical relevance, because flecainide has been shown to prolong ventricular monophasic action potential duration [Citation8] and the QT interval [Citation8,Citation9] in cardiac patients. A decrease in IKr is likely to contribute to the reduced capacity to develop appropriate shortening in APD90 upon a sudden reduction in cardiac cycle length, which would be manifested in decreased magnitude of APD90 change, or prolonged time required to achieve a steady-state reduction in APD90 value, or both. This assumption is fully supported by changes revealed in flecainide-treated heart preparations used in the present study (). Furthermore, in support of the important role played by the reduced repolarization reserve in this setting, previous studies have demonstrated that adaptation of ventricular repolarization to a brief tachycardia is attenuated in human patients with long QT syndrome [Citation17] and in transgenic rabbits with loss-of-function mutation of the IK channel.[Citation20]
Importantly, flecainide treatment was associated with a 2.5-fold increase in the risk of sudden arrhythmic death in patients with healed myocardial infarction enrolled in the CAST.[Citation5] In animal studies, arrhythmogenic responses to flecainide were attributed to drug-induced conduction abnormalities in remodeled ventricular muscle [Citation21] and impaired spatial repolarization gradients and epicardial activation-to-repolarization coupling.[Citation22,Citation23] The present study adds to understanding the mechanisms of flecainide-induced arrhythmia by showing for the first time that this agent may attenuate rate adaptation of ventricular repolarization. A protracted rate adaptation of ventricular repolarization has been considered as an index of increased arrhythmic susceptibility in clinical conditions associated with abrupt changes in cardiac activation rate, for example, during exercise,[Citation4] spontaneous onset and termination of atrial fibrillation,[Citation24] or ventricular pacing upon cardiac electrophysiological testing.[Citation25] The mechanism by which the impaired rate adaptation may promote arrhythmia is related to facilitation of the transient early afterdepolarization activity in ventricular cells with inappropriately prolonged action potential duration. These changes are likely to account for clinical observations suggesting that arrhythmogenic responses to flecainide are often precipitated by heart rate acceleration during exercise testing.[Citation10,Citation11]
Finally, this study has shown that during programmed ventricular stimulation, APD90-prolonging effect evoked by flecainide both in regular and extrasystolic beats tends to be greater while using a longer cycle length in the basic drive train (S1=550 ms versus S1-S1=300 ms). Attenuation of the flecainide effect at shorter pacing intervals could be potentially attributed to K+ accumulation in extracellular clefts upon fast cardiac activation rate, or incomplete deactivation of IKs, the slow component of the delayed rectifier.[Citation17] Both changes may contribute to increased net outward K+ current and hence to oppose flecainide effect on action potential duration. However, the assessment of the role played by these mechanisms in the rate-dependency of flecainide-induced prolongation of APD90 requires further electrophysiological studies.
Conclusions
In perfused guinea-pig hearts, flecainide attenuates adaptation of ventricular repolarization to sudden changes in cardiac cycle length provoked by sustained cardiac acceleration or extra-systolic beats. These findings contribute to better understanding of the mechanisms of flecainide-induced arrhythmia in clinical conditions associated with transient alterations in cardiac activation rate.
Declaration of interest
This study was supported by the Novo Nordisk Foundation. The author has no conflicts of interest.
References
- Locati EH, Maison-Blanche P, Dejode P, et al. Spontaneous sequences of onset of torsade de pointes in patients with acquired prolonged repolarization: Quantitative analysis of Holter recordings. J Am Coll Cardiol. 1995;25:1564–1575.
- Panidis IP, Morganroth J. Sudden death in hospitalized patients: Cardiac rhythm disturbances detected by ambulatory electrocardiographic monitoring. J Am Coll Cardiol. 1983;2:798–805.
- Denker S, Lehmann MH, Mahmud R, et al. Facilitation of macroreentry within the His-Purkinje system with abrupt changes in cycle length. Circulation. 1984;69:26–32.
- Gill JS, Baszko A, Xia R, et al. Dynamics of the QT interval in patients with exercise-induced ventricular tachycardia in normal and abnormal hearts. Am Heart J. 1993;126:1357–1363.
- Echt DS, Liebson PR, Mitchell B, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. New Engl J Med. 1991; 324:781–788.
- Follmer CH, Colatsky TJ. Block of delayed rectifier potassium current, IK, by flecainide and E-4031 in cat ventricular myocytes. Circulation. 1990;82:289–293.
- Wang DW, Kiyosue T, Sato T, Arita M. Comparison of the effects of class I antiarrhythmic drugs, cibenzoline, mexiletine and flecainide, on the delayed rectifier K+ current of guinea-pig ventricular myocytes. J Mol Cell Cardiol. 1996;28:893–903.
- Olsson SB, Edvardsson N. Clinical electrophysiologic study of antiarrhythmic properties of flecainide: Acute intraventricular delayed conduction and prolonged repolarization in regular paced and premature beats using intracardiac monophasic action potentials with programmed stimulation. Am Heart J. 1981;102:864–871.
- Lim KS, Jang IJ, Kim BH, et al. Changes in the QTc interval after administration of flecainide acetate, with and without co-administered paroxetine, in relation to cytochrome P450 2D6 genotype: data from an open-label, two-period, single-sequence crossover study in healthy Korean male subjects. Clin Ther. 2010;32:659–666.
- Hoffmann A, Wenk M, Follath F. Exercise-induced ventricular tachycardia as a manifestation of flecainide toxicity. Int J Cardiol. 1986;11:353–355.
- Anastasiou-Nana MI, Anderson JL, Stewart JR, et al. Occurrence of exercise-induced and spontaneous wide complex tachycardia during therapy with flecainide for complex ventricular arrhythmias: A probable proarrhythmic effect. Am Heart J. 1987;113:1071–1077.
- Osadchii OE, Woodiwiss AJ, Norton GR. Contractile responses to selective phosphodiesterase inhibitors following chronic beta-adrenoreceptor activation. Pflugers Arch. 2006;452:155–163.
- Osadchii OE, Olesen SP. Electrophysiological determinants of hypokalemia-induced arrhythmogenicity in the guinea-pig heart. Acta Physiol (Oxf). 2009;197:273–287.
- Osadchii OE. Dofetilide promotes repolarization abnormalities in perfused guinea-pig heart. Cardiovasc Drugs Ther. 2012;26:489–500.
- Conard GJ, Ober RE. Metabolism of flecainide. Am J Cardiol. 1984;53:41B–51.
- Pueyo E, Husti Z, Hornyik T, et al. Mechanisms of ventricular rate adaptation as a predictor of arrhythmic risk. Am J Physiol Heart Circ Physiol. 2010;298: H1577–H1587.
- Viskin S, Postema PG, Bhuiyan ZA, et al. The response of the QT interval to the brief tachycardia provoked by standing: A bedside test for diagnosing long QT syndrome. J Am Coll Cardiol. 2010;55:1955–1961.
- Eisner DA, Dibb KM, Trafford AW. The mechanism and significance of the slow changes of ventricular action potential duration following a change of heart rate. Exp Physiol. 2009;94:520–528.
- Paul AA, Witchel HJ, Hancox JC. Inhibition of the current of heterologously expressed HERG potassium channels by flecainide and comparison with quinidine, propafenone and lignocaine. Br J Pharmacol. 2002;136:717–729.
- Bentzen BH, Bahrke S, Wu K, et al. Pharmacological activation of Kv11.1 in transgenic long QT-1 rabbits. J Cardiovasc Pharmacol. 2011;57:223–230.
- Coromilas J, Saltman AE, Waldecker B, et al. Electrophysiological effects of anisotropic conduction and reentry in infracted canine hearts. Circulation. 1995;91:2245–2263.
- Osadchii OE, Soltysinska E, Olesen SP. Na+ channel distribution and electrophysiological heterogeneities in guinea-pig ventricular wall. Am J Physiol Heart Circ Physiol. 2011;300:H989–1002.
- Osadchii OE. Flecainide-induced proarrhythmia is attributed to abnormal changes in repolarization and refractoriness in perfused guinea-pig heart. J Cardiovasc Pharmacol. 2012;60:456–466.
- Grom A, Faber TS, Brunner M, et al. Delayed adaptation of ventricular repolarization after sudden changes in heart rate due to conversion of atrial fibrillation. A potential risk factor for proarrhythmia? Europace. 2005;7:113–121.
- Bueno-Orovio A, Hanson BM, Gill JS, et al. Slow adaptation of ventricular repolarization as a cause of arrhythmia? Methods Inf Med. 2014;53:320–323.