
Abstract
Objectives. This 24-week, multicenter, double-blind, randomized, placebo-controlled study (NCT00791999) compared efficacy and safety of certolizumab pegol (CZP) in combination with methotrexate (MTX) vs placebo plus MTX in Japanese rheumatoid arthritis (RA) patients with inadequate response to MTX.
Methods. In total, 316 patients were randomized 1:1:1:1 to subcutaneous CZP 100, 200, or 400 mg (induction dose: 200 mg or 400 mg CZP at Weeks 0, 2, and 4) plus MTX or placebo plus MTX every 2 weeks. Primary endpoint was ACR20 response at Week 12.
Results. ACR20 response rates were 62.5%, 76.8%, 77.6%, and 28.6% at Week 12, and 61.1%, 73.2%, 71.8%, and 24.7% at Week 24 for CZP 100, 200, and 400 mg, and placebo groups, respectively, with statistical significance between each CZP group and placebo. Change in Total Sharp Score over 24 weeks was significantly smaller in CZP 200 and 400 mg groups vs placebo. Improvements in health-related quality of life (HRQoL) were observed in all three CZP groups vs placebo. Incidence of adverse events was similar between CZP groups.
Conclusions. CZP plus MTX resulted in rapid, sustained reductions in RA signs and symptoms in Japanese patients with inadequate response to MTX, with significant inhibition of radiographic progression and improved HRQoL.
Introduction
Tumor necrosis factor alpha (TNF-α) inhibitors have significant beneficial effects on the signs and symptoms of rheumatoid arthritis (RA), the progression of joint damage, and on physical functioning and health-related quality of life (HRQoL), as shown in randomized controlled clinical trials [Citation1–8]. As biologic agents, TNF inhibitors are recommended either as monotherapy or in combination with non-biologic agents for RA patients responding inadequately to synthetic disease-modifying antirheumatic drugs (DMARDs) including methotrexate (MTX) [Citation9,Citation10].
Certolizumab pegol (CZP) is a PEGylated Fc-free anti-TNF-α agent. The RA Prevention of Structural Damage (RAPID) 1 and RAPID 2 international studies demonstrated that CZP plus MTX significantly improved the signs and symptoms of RA in patients with active disease despite MTX treatment [Citation5,Citation8]. The objective of the Japan RAPID (J-RAPID) study presented here was to investigate the efficacy and safety of CZP with concomitant MTX therapy in Japanese patients with active RA who have failed to respond adequately to MTX.
Materials and methods
Study overview
J-RAPID was a 24-week, phase 2/3 multicenter, double-blind, randomized placebo-controlled study (NCT00791999), conducted between 19 November 2008 and 18 August 2010 in 67 centers across Japan, in which patients with active RA and an inadequate response to MTX received CZP or placebo while continuing to take their previous dosage of MTX. The MTX regimen could not be changed after initiation of the study treatment.
Patients were randomized 1:1:1:1 to subcutaneous CZP 100, 200, or 400 mg plus MTX, or saline placebo plus MTX, every 2 weeks (Q2W). Block randomization was used to allocate patients to treatment arms. The random allocation sequence was generated using uniform random numbers from SAS® RANUNI function. The study drug allocation center was responsible for preparation and storage of the randomization table, study drug allocation, and confirmation of indistinguishability of study drugs, while the registration center was responsible for assignment of study drug numbers to patients. Patients randomized to CZP plus MTX received induction doses of 200 mg (100 mg group) or 400 mg (200 and 400 mg groups) at Weeks 0, 2 and 4. Patients randomized to placebo received an equivalent injection regimen of saline solution to maintain blinding. Study drug administration was performed by non-blinded personnel who were not allowed to engage in any other study activities. The MTX regimen could not be changed after initiation of the study treatment. The dose of MTX was set to 6–8 mg/week in accordance with the approved dose in Japan at the time of the clinical trial.
Patients who did not achieve an ACR20 response (i.e., ≥ 20% improvement according to the criteria of the American College of Rheumatology [ACR] [Citation11]) at Weeks 12 and 14 (ACR20 non-responders) were withdrawn from the study at Week 16 and were eligible to enter an open-label extension study thereafter.
The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and with the Pharmaceutical Affairs Law Standards for the Conduct of Clinical Trials on Drugs (Ministry of Health, Labour and Welfare Ordinance no. 28, 27 March 1997) and related notifications. Institutional review board approval was obtained at all centers and written informed consent was provided by all patients.
Patients
Since the international RAPID 2 study was used as a bridging model, J-RAPID adopted the same inclusion criteria, including disease duration, with some exceptions.
Eligible patients were aged from 20–74 years and had a diagnosis of RA defined by ACR (1987) criteria [Citation12] for 0.5–15 years. Disease duration was set in accordance with the criteria in the RAPID 2 study, whereas the lower age limit differed among the two studies, 18 years in the RAPID 2 study and 20 years in the present study, set in order to avoid including minors in the study. In addition, from a safety perspective, an upper limit of up to age 75 was set. Patients must have had active RA as defined by at least nine tender and nine swollen joints (among 68 and 66 joints of ACR definition, respectively) at screening and baseline, and have satisfied at least one of the following criteria at screening: erythrocyte sedimentation rate (ESR) of ≥ 30 mm/hour or C-reactive protein (CRP) of ≥ 1.5 mg/dL.
Patients must have received treatment with MTX (with or without folic acid) for 6 or more months before study drug administration, with the MTX dose fixed for 2 or more months beforehand and within the range of 6–8 mg/week.
Patients were excluded if they had received any biologic therapy for RA within the 6 months preceding the study (3 months for etanercept) or had received any investigational drug in the preceding 3 months. Patients who had received previous treatment with ≥ 2 TNF inhibitors or who had not initially responded to previous TNF inhibitor therapy were also excluded, as were those who had previously shown severe hypersensitivity or had an anaphylactic reaction. Oral corticosteroids (up to 10 mg/day prednisone equivalent), nonsteroidal anti-inflammatory drugs and selective cyclooxygenase-2 inhibitors were permitted as long as doses had been stable for ≥ 28 and ≥ 14 days, respectively, preceding study entry. Use of parenteral corticosteroids, intra-articular hyaluronic acid, azathioprine, cyclosporine, or DMARDs (with the exception of MTX) was not permitted within the 28 days before study Day 1. Patients were also excluded if they had history of demyelinating or convulsive disease of the central nervous system (e.g., multiple sclerosis and epilepsy), New York Heart Association Class III or IV congestive heart failure, infectious disease, hepatitis B or hepatitis C, malignant tumor or lymphoproliferative disorder, including lymphoma or signs and symptoms suggestive of lymphoproliferative disease. Patients with any indication of current or past tuberculosis as shown by clinical history, chest X-ray and/or positive tuberculin reaction test were also excluded unless preventive therapy by isoniazid was first taken.
Study assessments
After a screening period of 1–4 weeks prior to randomization, efficacy assessments were carried out over the 24-week treatment period as follows: at baseline, Weeks 1, 2, 4, 6, 8, 12, 14, 16, 20 and 24 or time of discontinuation. Safety was assessed at every visit. The primary efficacy endpoint was ACR20 response rate at Week 12. The key secondary endpoint was ACR20 response rate at Week 24.
Additional endpoints were: ACR20 response at other time points, and ACR50 and ACR70 response rates; individual ACR core component scores: number of tender joints, number of swollen joints, assessment of physical function using Health Assessment Questionnaire Disability Index (HAQ-DI), patient assessment of arthritis pain using Visual Analog Scale (VAS), patient global assessment of disease activity using VAS, physician global assessment of disease activity using VAS, CRP, and ESR; prevention of progression of joint destruction (change in van der Heijde modified Total Sharp Score [mTSS]) at Week 24; duration of morning stiffness; Disease Activity Score 28-joint assessment with ESR (DAS28[ESR]) remission rates and European League Against Rheumatism (EULAR) response [Citation13].
Radiographic assessments were performed at baseline and at Week 24 or at discontinuation using the mTSS [Citation14,Citation15]. The degree of joint erosion was assessed in 44 joints and joint space narrowing (JSN) in 42 joints. Radiographs were independently evaluated by two experienced assessors who were blinded to the treatment regimen and timing of radiography. Mean scores across two radiographic readers were used for analysis. Erosions and JSN were summed to obtain the mTSS. mTSS non-progression was defined as change from baseline in mTSS ≤ 0.5 units at Week 24.
HRQoL was assessed at baseline, Week 12, and Week 24 using the Short Form-36 Health Survey (SF-36), including the eight domains of Physical Functioning, Role Physical, Bodily Pain, General Health, Vitality, Social Functioning, Role Emotional and Mental Health, scored from 0 to 100 [Citation16].
Plasma samples were analyzed for determination of concentrations of CZP and anti-CZP antibody at every visit to Week 8, then at Weeks 12 and 24 or the time of discontinuation. Safety was assessed at all visits and at 12-week follow-up, and included adverse events (AEs), laboratory findings, body weight and vital signs, 12-lead ECGs and radiography of the chest. Serious AEs (SAEs) were those that resulted in death, were life-threatening, required or prolonged hospitalization, or resulted in significant disability or incapacity or were congenital anomalies/birth defects.
Statistical analysis
The sample size was based on an expected ACR20 response rate of 22% in the placebo group and ≥ 50% in the 200 mg and 400 mg groups, as per previous clinical experience with CZP. Verification of superiority of the 200 mg and 400 mg doses over placebo for the primary endpoint would then have 90% power at a two-sided significance level of 2.5% in order to preserve the overall Type I error rate at α = 0.05, with 71 patients per group (300 overall, pre-randomization).
The primary population for analysis of the primary endpoint was the full analysis set (FAS) of patients who received ≥ 1 dose of study drug and provided ≥ 1 efficacy data thereafter. The safety population contained all patients who received ≥ 1 dose of study drug.
ACR responses were determined using non-responder imputation. Patients who violated study protocol, received rescue medication, or withdrew for any reason were considered non-responders from that time point onward.
Logistic regression was used for ACR response comparisons. Changes in ACR core parameters and changes in total tender and swollen joint scores between baseline and Week 24 were examined using analysis of covariance (ANCOVA) with the baseline value as the covariate, and using last observation carried forward (LOCF) imputation for missing data. Changes from baseline to the assessment time point for DAS28(ESR), SF-36 and duration of morning stiffness, were also analyzed using ANCOVA (LOCF) with treatment group as a factor and the baseline value as a covariate. For EULAR response (good, moderate or no response), intergroup comparisons using logistic regression at each time point were carried out using LOCF imputation.
Radiographic outcomes were summarized for those patients with complete radiographic data available for two assessment points (at baseline and post-drug administration). mTSS values at Week 24 were estimated employing linear extrapolation in patients in whom administration was discontinued before Week 24 using values obtained at the discontinuation visit. mTSS values at Week 24 for early withdrawals were estimated by linear extrapolation of the last available value to Week 24, assuming disease progression occurred at the same rate between baseline and withdrawal, as commonly used for missing mTSS data [Citation5,Citation17,Citation18]. In order to examine the change in rank from baseline, ANCOVA on the ranks was performed with treatment as factors and rank baseline mTSS as covariate.
Treatment-emergent AEs (TEAEs) included all events from after the administration of the study drug until the last evaluation visit (not including the safety follow-up visit). TEAEs were coded by system organ class and preferred term using MedDRA terminology (v11.1).
Results
Patient characteristics and disposition
A total of 316 patients (FAS) were randomized () to CZP 100 mg (n = 72), CZP 200 mg (n = 82), CZP 400 mg (n = 85) or placebo (n = 77). In the CZP 100, 200 and 400 mg groups, 51, 66 and 65 patients completed 24 weeks of double-blind treatment, respectively, compared with 25 patients in the placebo group. There were no marked differences in the patient background characteristics or severity of the disease at baseline among the four groups ().
Figure 1. Patient disposition. aACR20 response was not achieved at Weeks 12 and 14; bEfficacy of study drug was insufficient at times other than at Weeks 12 and 14. Patients not showing an ACR20 response at Weeks 12 and 14 were withdrawn from the study at Week 16 and were eligible to enter an open-label extension, as were patients completing the study.
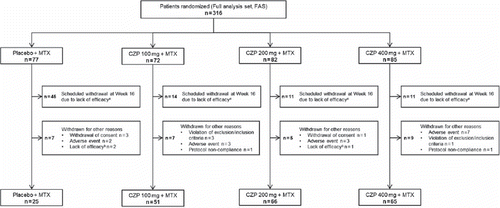
Table 1. Patient demographics and disease status at baseline (FAS population).
More placebo plus MTX-treated patients withdrew at Week 16 due to a lack of ACR20 response at Weeks 12 and 14 relative to the CZP 100 mg, 200 mg and 400 mg plus MTX groups; fewer patients randomized to placebo plus MTX completed 24 weeks’ treatment than in the CZP 100 mg 200 mg, and 400 mg plus MTX groups ().
Clinical efficacy
All active treatment groups showed higher ACR responses at Weeks 12 and 24 compared to placebo plus MTX ( and ). The superiority of all CZP plus MTX groups compared to placebo plus MTX in ACR20 response was statistically significant at Weeks 12 and 24 (p < 0.0001) ( and ).
Figure 2. ACR response rates and improvements in DAS28(ESR) and HAQ-DI scores up to Week 24: (a) ACR20, ACR50, and ACR70 response rates at Week 12, (b) ACR20, ACR50, and ACR70 response rates at Week 24, (c) ACR20 response over time (FAS population; NRI: patients who received rescue medication, or who withdrew for any reason, were considered non-responders from that time point forward), (d) Improvements in DAS28(ESR) over time (FAS population; LOCF), (e) Improvements in HAQ-DI over time (FAS population; LOCF).
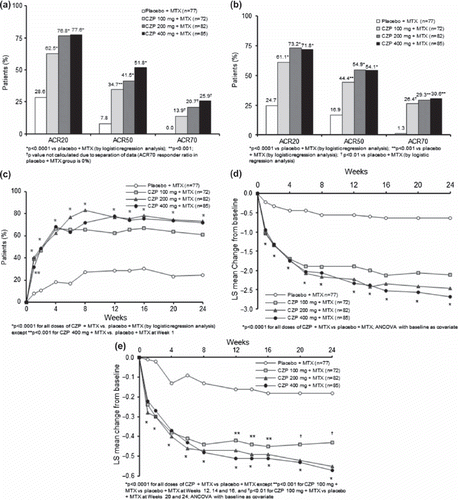
ACR50 responses were statistically significantly higher with all doses of CZP at Weeks 12 and 24 compared to those of placebo, and ACR70 responses were significantly higher at Week 24 ( and ). Statistical analysis could not be performed for ACR70 at Week 12 because of the 0% response in the placebo group. ACR responses in the 100 mg CZP group were less pronounced compared to those of the higher dose groups. The onset of response with CZP plus MTX was rapid across groups, with a significantly improved ACR20 response compared to placebo reported from Week 1 (p < 0.001). For all doses, the ACR20 response had peaked by Week 8 and was sustained to Week 24 (). ACR50 responses were also rapid, with statistical significance reported from Week 2 with 100 mg and 200 mg doses, and all doses were significantly better than placebo at Week 4 (data not shown).
CZP plus MTX treatment was also associated with statistically significant improvements in all ACR core components at Weeks 12 and 24 (). This was statistically significant from Week 1 for all CZP doses compared to placebo for all ACR components (p < 0.001). A marked improvement in DAS28(ESR) was observed with all doses of CZP plus MTX from Week 1 (p < 0.0001) and sustained throughout the trial (). DAS28(ESR) remission rates (DAS28(ESR) < 2.6) were also markedly higher with CZP than with placebo plus MTX at Week 12 (8.3%, 16.0% and 11.8% for CZP 100 mg, 200 mg and 400 mg plus MTX, respectively, vs 0% for placebo plus MTX) and at Week 24 (20.8%, 17.1% and 25.9%, vs 0% placebo plus MTX).
Table 2. Least squares (LS) mean change from baseline or ratio of geometric mean to BL at Weeks 12 and 24 in ACR core components and other endpoints (FAS population with LOCF).
Moderate or good EULAR responses were reported in 76.4%, 86.4% and 90.6% of patients receiving CZP 100 mg, 200 mg and 400 mg plus MTX, respectively, compared with 36.4% of patients on placebo plus MTX at Week 12 and 77.8%, 85.4% and 89.4% vs 29.9% at Week 24.
Physical function and HRQoL
HAQ-DI was significantly improved with all doses of CZP plus MTX at Week 12 and Week 24 (p < 0.0001, except for CZP 100 mg at Week 12 p < 0.001 and Week 24 p < 0.005) (). These improvements were significant from Week 1 and sustained at each visit throughout the trial ().
Least squares mean changes in SF-36 scores showed the beneficial effects of CZP plus MTX on HRQoL. Changes in physical component summary scores from baseline were significant for all CZP groups compared to placebo at Week 12 and sustained at Week 24 (). Similarly, changes in mental component summary scores were significant at Week 12 for the 200 and 400 mg CZP groups compared to those of placebo and maintained at Week 24 ().
Inhibition of progression of structural damage
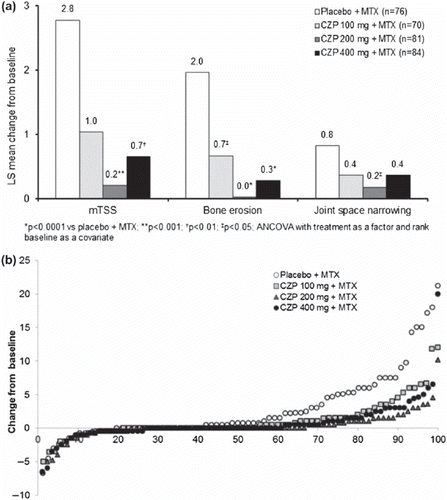
Radiographic data were available at both baseline and after drug administration for 76 of 77 placebo patients and 70 of 72, 81 of 82, and 84 of 85 patients in the CZP 100, 200, and 400 mg groups, respectively. CZP plus MTX inhibited the progression of structural damage compared to placebo at Week 24 ( and ). Mean changes from baseline to Week 24 in total mTSS were significantly lower in the CZP 200 mg (p < 0.001) and CZP 400 mg (p < 0.01) plus MTX groups compared to placebo. The progression in joint erosion was also significantly lower in all CZP groups compared to placebo. The mean change in JSN was significantly lower for the CZP 200 mg dose compared to placebo. Numerically, across all three radiographic measures the greatest inhibition in the progression of joint damage was seen with CZP 200 mg plus MTX (). A higher percentage of patients who received CZP 200 mg or 400 mg plus MTX achieved mTSS non-progression at Week 24 than with placebo plus MTX (47.4% of patients on placebo plus MTX compared with 74.1% (p < 0.001) and 70.2% (p < 0.01) of patients receiving CZP 200 mg and 400 mg plus MTX, respectively). Linear extrapolation was used to impute missing radiographic data in 51/76 placebo patients (including those who withdrew at Week 16) and 21/70, 15/81 and 20/84 CZP 100, 200 and 400 mg patients, respectively.
Figure 3. Inhibition of progression of structural damage: (a) Change from baseline in total mTSS, bone erosion and joint space narrowing at Week 24 (FAS population, linear extrapolation). *p < 0.0001 vs placebo + MTX; **p < 0.001; †p < 0.01; ‡p < 0.05; ANCOVA with treatment as a factor and rank baseline as a covariate, (b) Cumulative probability plot of change from baseline in mTSS at Week 24 (FAS population, linear extrapolation).
Safety
TEAEs were similar between treatment groups, as reported in . The majority of events were of mild to moderate severity. SAEs were infrequent, occurring in 13 patients: 3 (4.2%), 4 (4.9%), 5 (5.9%) and 1 (1.3%) for CZP 100 mg, 200 mg and 400 mg plus MTX and placebo plus MTX, respectively (). There were no cases of tuberculosis or malignant disease and no deaths during these 24 weeks of the study.
Table 3. Treatment-emergent adverse events (safety population).
The most frequent AE by system organ class was infections and infestations, occurring in 84 patients (35.1%) in the CZP-treated groups and 19 patients (24.7%) in the placebo group. Administration site reaction (2.5% and 0.0%), injection site erythema (0.8% and 0.0%), injection site hematoma (0.4% and 0.0%), injection site hemorrhage (0.0% and 1.3%), injection site mass (0.4% and 0.0%) and injection site reaction (0.4% and 1.3%) were reported in CZP-treated groups and placebo group, respectively, and the majority of reactions were mild. In the CZP groups, the proportions of patients who showed high aspartate aminotransferase and alanine aminotransferase levels after administration despite normal values at baseline were slightly higher than in the placebo group.
Anti-CZP antibodies were noted in 11 patients; 9 (12.5%) were in the 100 mg group and 1 each (1.2%) in the 200 and 400 mg groups, which may have partially contributed to the lower efficacy in the 100 mg group compared with the higher doses (Week 24 ACR20 response rate: CZP 100 mg, 61.1%; CZP 200 mg, 73.2%; and CZP 400 mg, 71.8%). There were no differences in the safety profile between patients with and without anti-CZP antibodies.
Discussion
In this study, treatment with CZP plus MTX was associated with a rapid and significant reduction in the signs and symptoms of RA, inhibition of the progression of joint damage, and improved physical function and HRQoL in Japanese patients with active RA and an inadequate response to MTX.
The primary efficacy endpoint, ACR20 response at Week 12, was met by significantly more patients in the CZP 100, 200 and 400 mg groups compared to those in the placebo group. Higher responses vs placebo plus MTX were obtained with CZP plus MTX within the first week of treatment, and were sustained to Week 24. These differences were statistically significant. Rapid and sustained effects of active treatment were evident for all ACR core components, indicated by the effects on tender and swollen joint counts, on physical functioning (HAQ-DI) and on the patient's perception of pain, similar to other clinical trials [Citation5,Citation7,Citation8,Citation19]. Overall HRQoL significantly improved with all doses of CZP plus MTX; improvement in a greater number of individual subscales was seen with the two higher doses.
Both CZP 200 mg and CZP 400 mg plus MTX groups demonstrated statistically significant inhibition of joint damage vs placebo. Furthermore, CZP 200 mg plus MTX showed numerically better efficacy and greater inhibition of joint damage than CZP 100 mg plus MTX, and no additional benefit of the higher CZP 400 mg plus MTX dose over the 200 mg plus MTX dose was observed. These findings support the results of the previous Phase III studies, and confirm 200 mg Q2W as the optimum dose of CZP [Citation5,Citation8].
The CZP 100 mg Q2W group was included in the study on the assumption that the average body weight of the Japanese patients may be lower than that in the patients enrolled in the international studies. Consequently, the efficacy of the lower dose (CZP 100 mg) was compared to the standard CZP 200 mg Q2W dose, and was found to give lower ACR20, ACR50 and ACR70 response rates at both Weeks 12 and 24 (), as well as less inhibition of joint damage at Week 24 (). In addition, subgroup analysis revealed that the efficacy of CZP was not associated with the patients’ body weight (data not shown).
There were slight differences in the baseline history of TNF inhibitor usage between the placebo and CZP 100 mg groups (7/77 patients in the CZP 100 mg group vs 15/72 patients in the placebo group). In subgroup analyses, ACR20 response rates were higher in the CZP groups than in the placebo group, regardless of prior TNF inhibitor exposure (data not shown), therefore it is presumed that the effect of previous TNF inhibitor usage on ACR20 response rate was limited.
The difference in the efficacy between dosing regimen of CZP 400 mg at Weeks 0, 2, and 4 (: CZP 200 mg group and 400 mg group) and CZP 200 mg at Weeks 0, 2, and 4 (: CZP 100 mg group) was not very clear. In order to investigate the additive effect of induction doses on ACR20 response rate in Japanese RA patients, simulation of two dosing regimens was performed, (1) with induction dose (CZP 400 mg at Weeks 0, 2 and 4, followed by 200 mg Q2W) and (2) without induction dose (CZP 200 mg Q2W from Week 0), using a pharmacokinetic (PK) model and a PK/pharmacodynamic (PD) model. The results predicted that use of the induction doses improves the efficacy of CZP during the initial stage of treatment, which peaks at Weeks 7 and 8. The median ACR20 response rates at the peak time points were around 68% and 69% with the induction doses vs around 56% and 57% without the induction doses, suggesting approximately 10% increase in ACR20 is predicted by administering the induction dose. The PK/PD model predicts that the beneficial effect of the induction dose will remain apparent at Week 12.
CZP plus MTX was well-tolerated at all three doses assessed, with low rates of discontinuation due to AEs, with the CZP 100 mg and 200 mg groups showing similar rates to the placebo group. Most events were of mild to moderate severity and SAEs were infrequent, occurring in 4.2–5.9% of CZP patients. Overall, there were no differences in the safety profile among the doses in the CZP-treated groups. Although the rates of AEs were higher compared to RAPID 1 [Citation8] and RAPID 2 [Citation5], this was true for both the placebo and the CZP-treated patients. The safety profile of CZP was similar to the results of previous clinical studies, as expected for anti-TNF agents. There was no new event in terms of safety in these Japanese patients in comparison with the international data.
A relatively high incidence of tuberculosis in Japan (24.8 cases per 100,000 per year, five times the US incidence [Citation20]) has given rise to concerns over the possibility of increased rates of this infection with TNF inhibitor therapy [Citation21]. However, in the present study there were no reports of tuberculosis. Although confirmation needs to be obtained through postmarketing surveillance studies, the findings herein are concordant with such data for infliximab [Citation20] and etanercept [Citation22], where early fears over increased risk of serious respiratory infection in Japanese patients specifically were shown to be unfounded.
Limitations of this study exist due to the specific design herein; patients with history of use of ≥ 2 TNF inhibitors and those who had not previously responded to TNF inhibitor therapy were excluded. Consequently, the results of this study may not accurately reflect treatment of CZP with MTX in patients with these profiles. Likewise, as the study was only 24 weeks in length, the safety and efficacy of CZP will need further assessment during the long-term open-label extension of this study.
Adoption of a tight control approach to treatment, through regular patient follow-up with appropriate adaptation of therapy, to reach a state of low disease activity within 3–6 months of treatment is among the key recommendations of an international task force examining the treat-to-target approach in RA [Citation9]. A treatment regimen with a rapid response may facilitate early decision making and enable the treatment of non-responding patients to be optimized quickly in line with these recommendations [Citation23]. A post-hoc analysis of the international RAPID 1 study demonstrated that the majority of DAS28 response with CZP (DAS28 improvement of ≥ 1.2) was reported within 12 weeks of initiating treatment, and that early DAS28 non-response is a predictor of poorer long-term outcomes [Citation24]. In this study with Japanese patients, reduction in the signs and symptoms of RA by treatment with CZP plus MTX was similarly rapid, and significant improvement was achieved at Week 12, suggesting the possibility of early prediction of response at Week 12. This will be further examined using long-term data from the open-label extension of this trial.
Acknowledgments
The authors acknowledge ‘Matladi Ndlovu, PhD, UCB Pharma, Brussels, Belgium, and Yumiko Wada, PhD, UCB Japan, Tokyo, Japan for publication management and Costello Medical Consulting for editorial and administrative support.
The authors also acknowledge all investigators from the J-RAPID study:
Hokkaido University Hospital (T. Atsumi), Hokkaido Medical Center for Rheumatic Diseases (K. Tanimura), Tomakomai City Hospital (A. Fujisaku), Oki Medical Clinic (I. Oki), Tohoku KoseiNenkin Hospital (T. Kodera), East-Sendai Rheumatism and internal Medicine Clinic (T. Izumiyama), Taga General Hospital (S. Ota), Inoue Hospital (H. Inoue), Saitama Medical Center (K. Amano), Jichi Medical University Hospital (T. Yoshio), Tsukuba University Hospital (T. Sumida), Chiba University Hospital (N. Watanabe), Yonemoto Orthopedic Clinic (K. Yonemoto), The University of Tokyo Hospital (K. Kawahata), Tokyo Medical and Dental University Hospital Faculty of Medicine (H. Kohsaka), Institute of Rheumatology Tokyo Women's Medical University (H. Yamanaka), Keio University Hospital (M. Kuwana), Jikei University Hospital (D. Kurosaka), Juntendo University Hospital (Y. Takasaki), Showa University Hospital (T. Kasama), Toho University Ohashi Medical Center (T. Ogawa), Kyorin University Hospital (Y. Arimura), Fukuhara Hospital (A. Yamaguchi), Tokyo Metropolitan Bokutoh Hospital (M. Nagashima), Tokyo Metropolitan Ohtsuka Hospital (T. Yamada), Yokohama Rosai Hospital (Y. Kita), Tokai University Hospital (Y. Suzuki), Kitasato University Hospital (S. Hirohata), Sagamihara Hospital (T. Matsui), Gunma University Hospital (Y. Nojima), Nagano Red Cross Hospital (T. Kanamono), Nagoya University Hospital (T. Kojima), Nagoya Medical Center (A. Kaneko), Kondo Orthopedic and Rheumatology Clinic (K. Kondo), Ito Orthopedic Clinic (T. Ito), Nagoya City university Hospital (Y. Hayami), Niigata University Medical and Dental Hospital (M. Nakano), Niigata Rheumatic Center (A. Murasawa), Saiseikai Takaoka Hospital (S. Honjo), Osaka Minami Medical Center (Y. Saeki), Matsubara Mayflower Hospital (T. Matsubara), Okayama University Hospital (H. Wakabayashi), Higashi-Hiroshima Memorial Hospital (S. Yamana), Kurashiki Medical Clinic (Y. Yoshinaga), Hiroshima Clinic (K. Amano), Sanin Rosai Hospital (H. Kishimoto), Kagawa University Hospital (H. Dobashi), Tokushima University Hospital (J. Kishi), Matsuyama Red Cross Hospital (S. Mizuki), University of Occupational and Environmental Health, Hospital (Y. Tanaka), Kitakyushu Municipal Medical Center (H. Nishizaka), Kyushu Medical Center (H. Miyahara), Shono Rheumatism Clinic (E. Shono), Kondo Rheumatology and Orthopedic Clinic (M. Kondo), Kurume University Medical Center (T. Fukuda), St.Mary's Hospital (T. Nakano), Nagasaki University Hospital of Medicine and Dentistry (A. Kawakami), Nagasaki Medical Center (K. Migita), Sasebo Chuo Hospital (Y. Ueki), Nagasaki Medical Hospital of Rheumatology (M. Tsuboi), Nagasaki Atomic Bomb Hospital (M. Nakashima), Oribe Clinic of Rheumatism and Medicine (M. Oribe), Kumamoto Saishunso National Hospital (S. Mori), Kumamoto Orthopaedic Hospital (M. Sakaguchi), Shimin- No-Mori Hospital (T. Hidaka), Kagoshima Red Cross Hospital (T. Matsuda).
Conflicts of interest
The competing interests of all authors are provided below.
KY has served as a consultant for UCB Pharma, Pfizer, Abbott, BMS, Roche, Chugai, Mitsubishi-Tanabe and Eisai, and has received research funding from UCB Pharma, Pfizer, Abbott, Santen Mitsubishi-Tanabe, and Eisai.
TT has served as a consultant for AstraZeneca, Eli Lilly, Novartis, Mitsubishi-Tanabe and Asahi Kasei, and has received research support from Abott, Astellas, BMS, Chugai, Daiichi-Sankyo, Eisai, Janssen, Mitsubishi-Tanabe, Nippon Shinyaku, Otsuka, Pfizer, Sanofi-Aventis, Santen, Takeda and Teijin, and has served on speaker bureaus for Abbott, BMS, Chugai, Eisai, Janssen, Mitsubishi-Tanabe, Pfizer and Takeda.
HY has served as a consultant for, and received research funding from, UCB Pharma, Abbott, Astellas, BMS, Chugai, Eisai, Janssen, Mitsubishi-Tanabe, Pfizer and Takeda.
NI has received research funding from Takeda, Mitsubishi-Tanabe, Astellas, Chugai, Abbott, BMS, Eisai, Janssen, Kaken and Pfizer, and has served on speaker bureaus for Takeda, Mitsubishi-Tanabe, Astellas, Chugai, Abbott, BMS, Eisai, Janssen, Kaken, Pfizer, Taisho-Toyama and Otsuka.
YT has received research funding from BMS, MSD, Chugai, Mitsubishi-Tanabe, Astellas, Abbott, Eisai and Janssen, and has served on speaker bureaus for UCB Pharma, Mitsubishi-Tanabe, Abbott, Eisai, Chugai, Janssen, Santen, Pfizer, Astellas, Daiichi-Sankyo, GSK, AstraZeneca, Otsuka, Actelion, Eli Lilly, Nippon Kayaku, Quintiles Transnational and Ono.
KE has served as a consultant for UCB Pharma.
AW has received research support from Astellas, Daiichi- Sankyo, Kyorin, Shionogi, Taisho, Dainippon-Sumitomo, Taiho, Toyama Chemical and Meiji Seika, and has served on speaker bureaus for Abott, MSD, Otsuka, GSK, Shionogi, Daiichi-Sankyo, Taisho-Toyama, Dainippon-Sumitomo, Mitsubishi-Tanabe, Toyama Chemical, Bayer and Pfizer.
HO has served as a consultant for UCB Pharma and Astellas.
TS is an employee of Otsuka.
YS is an employee of UCB Pharma.
DvH has served as a consultant for, and received research support from, AbbVie, Amgen, AstraZeneca, BMS, Centocor, Chugai, Daiichi, Eli Lilly, GSK, Janssen, Merck, Novartis, Novo-Nordisk, Otsuka, Pfizer, Roche, Sanofi-Aventis, Schering-Plough, UCB Pharma and Vertex. DvH is also director of Imaging Rheumatology bv.
NM has received research support from Pfizer, Takeda, Mitsubishi-Tanabe, Chugai, Abbott, Eisai and Astellas.
TK has served on speaker bureaus for UCB Pharma, Pfizer, Chugai, Abbott, Mitsubishi-Tanabe, Takeda, Eisai, Santen, Astellas, Taisho-Toyama, BMS, Teijin and Daiichi-Sankyo.
References
- Maini R, St Clair EW, Breedveld F, Furst D, Kalden J, Weisman M, et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet. 1999;354(9194):1932–9.
- Weinblatt ME, Kremer JM, Bankhurst AD, Bulpitt KJ, Fleischmann RM, Fox RI, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340(4):253–9.
- Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA, et al. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2003;48(1):35–45.
- Maini RN, Breedveld FC, Kalden JR, Smolen JS, Furst D, Weisman MH, et al. Sustained improvement over two years in physical function, structural damage, and signs and symptoms among patients with rheumatoid arthritis treated with infliximab and methotrexate. Arthritis Rheum. 2004;50(4):1051–65.
- Smolen J, Landewe RB, Mease P, Brzezicki J, Mason D, Luijtens K, et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis. 2009;68(6):797–804.
- Emery P, Fleischmann R, van der Heijde D, Keystone EC, Genovese MC, Conaghan PG, et al. The effects of golimumab on radiographic progression in rheumatoid arthritis: results of randomized controlled studies of golimumab before methotrexate therapy and golimumab after methotrexate therapy. Arthritis Rheum. 2011;63(5):1200–10.
- Fleischmann R, Vencovsky J, van Vollenhoven RF, Borenstein D, Box J, Coteur G, et al. Efficacy and safety of certolizumab pegol monotherapy every 4 weeks in patients with rheumatoid arthritis failing previous disease-modifying antirheumatic therapy: the FAST4WARD study. Ann Rheum Dis. 2009;68(6):805–11.
- Keystone E, Heijde D, Mason D Jr., Landewe R, Vollenhoven RV, Combe B, et al. Certolizumab pegol plus methotrexate is significantly more effective than placebo plus methotrexate in active rheumatoid arthritis: findings of a fifty-two-week, phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum. 2008;58(11):3319–29.
- Smolen JS, Aletaha D, Bijlsma JW, Breedveld FC, Boumpas D, Burmester G, et al. Treating rheumatoid arthritis to target: recommendations of an international task force. Ann Rheum Dis. 2010;69(4):631–7.
- Mok CC, Tam LS, Chan TH, Lee GK, Li EK. Management of rheumatoid arthritis: consensus recommendations from the Hong Kong Society of Rheumatology. Clin Rheumatol. 2011;30(3):303–12.
- Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 1995;38(6):727–35.
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31(3):315–24.
- van Gestel AM, Prevoo ML, van 't Hof MA, van Rijswijk MH, van de Putte LB, van Riel PL. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism Criteria. Arthritis Rheum. 1996;39(1):34–40.
- van der Heijde D. How to read radiographs according to the Sharp/van der Heijde method. J Rheumatol. 2000;27(1):261–3.
- van der Heijde D, Simon L, Smolen J, Strand V, Sharp J, Boers M, et al. How to report radiographic data in randomized clinical trials in rheumatoid arthritis: guidelines from a roundtable discussion. Arthritis Rheum. 2002;47(2):215–8.
- Ware JE, Kosinski M. SF-36 Physical and Mental Health Summary Scales: Manual for Users Version 1. 2nd ed. Lincoln, Rhode Island, USA: Quality Metric; 2001.
- Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti–tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: A randomized, placebo–controlled, 52–week trial. Arthritis Rheum. 2004;50(5): 1400–11.
- Cohen SB, Keystone E, Genovese MC, Emery P, Peterfy C, Tak PP, et al. Continued inhibition of structural damage over 2 years in patients with rheumatoid arthritis treated with rituximab in combination with methotrexate. Ann Rheum Dis. 2010;69(6):1158–61.
- Strand V, Mease P, Burmester GR, Nikai E, Coteur G, van Vollenhoven R, et al. Rapid and sustained improvements in health-related quality of life, fatigue, and other patient-reported outcomes in rheumatoid arthritis patients treated with certolizumab pegol plus methotrexate over 1 year: results from the RAPID 1 randomized controlled trial. Arthritis Res Ther. 2009;11(6):R170.
- Takeuchi T, Tatsuki Y, Nogami Y, Ishiguro N, Tanaka Y, Yamanaka H, et al. Postmarketing surveillance of the safety profile of infliximab in 5000 Japanese patients with rheumatoid arthritis. Ann Rheum Dis. 2008;67(2):189–94.
- Komano Y, Tanaka M, Nanki T, Koike R, Sakai R, Kameda H, et al. Incidence and risk factors for serious infection in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitors: a report from the Registry of Japanese Rheumatoid Arthritis Patients for Longterm Safety. J Rheumatol. 2011;38(7):1258–64.
- Koike T, Harigai M, Inokuma S, Inoue K, Ishiguro N, Ryu J, et al. Postmarketing surveillance of the safety and effectiveness of etanercept in Japan. J Rheumatol. 2009;36(5):898–906.
- Keystone EC, Curtis JR, Fleischmann RM, Furst DE, Khanna D, Smolen JS, et al. Rapid improvement in the signs and symptoms of rheumatoid arthritis following certolizumab pegol treatment predicts better longterm outcomes: post-hoc analysis of a randomized controlled trial. J Rheumatol. 2011;38(6):990–6.
- van der Heijde D, Keystone EC, Curtis JR, Landewe RB, Schiff MH, Khanna D, et al. Timing and magnitude of initial change in disease activity score 28 predicts the likelihood of achieving low disease activity at 1 year in rheumatoid arthritis patients treated with certolizumab pegol: a post-hoc analysis of the RAPID 1 trial. J Rheumatol. 2012;39(7):1326–33.