Abstract
Heterocyclic and open-chain dipeptide-derived nitriles have been synthesized, containing an additional electrophilic center enabling the subsequent covalent modification of the thioimidate nitrogen formed in situ at the active site of the enzyme. The inhibitory potential of these nitriles against the cysteine proteases papain and cathepsins L, S, and K was determined. The open-chain dipeptide nitriles 8 and 10 acted as moderate reversible inhibitors, but no evidence for an irreversible inhibition of these enzymes was discernable.
| Abbreviations: | ||
| Ac-, | = | acetyl-; |
| CHAPS, | = | 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate; |
| dec, | = | decomposition; |
| DMF, | = | N,N-dimethyl formamide; |
| DMSO, | = | dimethyl sulfoxide; |
| DTT, | = | (±)-threo-2,3-dihydroxy-1,4-butanedithiol; |
| d, | = | doublet; |
| EDTA, | = | ethylenediamine tetraacetic acid; |
| EI, | = | electron impact ionization; |
| ESI, | = | electrospray ionization; |
| m, | = | multiplet; |
| Me, | = | methyl; |
| Mec, | = | 4-methylcoumarin-7-yl |
| mp, | = | melting point; |
| MS, | = | mass spectrometry; |
| n.d., | = | not determined; |
| neg, | = | negative mode; |
| NMR, | = | nuclear magnetic resonance; |
| Np, | = | para-nitrophenyl; |
| pos, | = | positive mode; |
| RDA, | = | retro-Diels-Alder fragmentation; |
| s, | = | singlet; |
| sat, | = | saturated; |
| t, | = | triplet; |
| THF, | = | tetrahydrofurane; |
| Z-, | = | benzyloxycarbonyl- |
Introduction
Among the large and heterogeneous class of cysteine proteases, the papain-like cysteine proteases have attracted remarkable interest in recent research, not only due to their vital role in various human physiological and pathological processes but also because of their importance as virulence factors in different human-pathogenic parasitesCitation1. Common characteristics of papain-like cysteine proteases are the active site thiol and the importance of the S2–P2 interaction as the main determinant for substrate specificity. In the field of inhibition of these enzymes by low-molecular weight synthetic substances, the focus is directed toward developing potent reversible inhibitors as they have a lower risk of side effects and toxicity during the long-term treatment of chronic diseases. However, compounds that act by irreversible covalent modification of the active site cysteine are of great value, as the inhibition of the enzyme is complete and insurmountableCitation2. Irreversible inhibitors are therefore attractive candidates for treating microbial infections and valuable tools for elucidating the in vivo functions of the enzyme. Compounds of different classes have been reported as irreversible inactivators of cysteine proteasesCitation3, including peptide-derived epoxidesCitation2,Citation4 and related aziridinesCitation5, diazoketonesCitation6, and Michael acceptors such as vinyl sulfones and peptidyl ene dionesCitation7–9. The inherent chemical reactivity of these agents limits their applicability due to unspecific reactions with other biomolecules.
The class of peptide nitriles has received considerable attention because they interact in a covalent-reversible manner with the thiol of the active site to form an enzyme-bound thioimidateCitation10. As the cyano group represents a soft electrophile, they are generally selective for cysteine proteases over other proteasesCitation11; however, exceptions have been observed for instance in the case of the serine protease dipeptidyl peptidase IVCitation12. To combine the benefits of the nitriles with those of irreversible inhibitors, we envisaged the development of compounds capable of facilitating an intramolecular “trapping” of the enzyme-bound thioimidate adduct. This irreversible “trapping” of the temporary thioimidate can be accomplished by the introduction of a nucleofuge in a suitable position relative to the cyano group. The attack of the thioimidate nitrogen then leads to the formation of a five (or six)-membered heterocyclic ring. 2-Alkylthio-4-oxo-quinazoline-3-acetonitriles and the analogous thieno[3,2-d]pyrimidine-3-acetonitriles () are molecules that react with thiols in this mannerCitation13. Such reactions, where the initial step generates the functionality for the following one, belong to the so called domino or cascade reactions. The particular conversion depicted in matches the anionic type of cascade reaction, according to the classification introduced by TietzeCitation14. As these nitriles are devoid of cysteine protease-inhibiting activity, probably due to the lack of structural elements that can be recognized by the enzyme’s subsites, it was intended to apply the concept to compounds of more peptidic character, which is reported herein.
Figure 1. Reaction of 2-alkylthio-4-oxo-quinazolineacetonitriles and analogous thieno[3,2-d]pyrimidineacetonitriles with aliphatic thiols to imidazo-fused tricyclic systems by trapping of thioimidates; X = CH=CH or SCitation13.
![Figure 1. Reaction of 2-alkylthio-4-oxo-quinazolineacetonitriles and analogous thieno[3,2-d]pyrimidineacetonitriles with aliphatic thiols to imidazo-fused tricyclic systems by trapping of thioimidates; X = CH=CH or SCitation13.](/cms/asset/77bc4898-f535-4cce-aca0-a60a7f7aca9e/ienz_a_379902_f0001_b.gif)
Materials and methods
General methods and materials
Melting points were determined on a Büchi 510 oil bath apparatus and are not corrected. Thin layer chromatography was performed on aluminum-supported plates of silica gel 60. Optical rotations were determined on a PerkinElmer 241 polarimeter. 1H NMR spectra (500 MHz) and 13C NMR spectra (125 MHz) were recorded on a Bruker Avance 500. Mass spectra were obtained on an API 2000 spectrometer from Applied Biosystems (ESI; sprayed from a 10−5 M solution in 2 mM NH4OAc/MeOH 1:1; volumetric flow rate 10 μL/min) and on an A.E.I. MS-50 spectrometer (EI; 70 eV). Enzymatic activity of papain and cathepsins was measured using a Varian Cary Bio 50 UV/Vis spectrophotometer and a PerkinElmer luminescence spectrometer LS 50 B, respectively. Papain was purchased from Sigma-Aldrich, Steinheim, Germany. Cathepsin L from bovine liver and recombinant human cathepsin S were purchased from Calbiochem, Darmstadt, Germany. Recombinant human cathepsin K (expressed in Pichia pastoris) was a gift from D. BrömmeCitation15. The substrates, Z-Phe-Arg-NHNp, Z-Phe-Arg-NHMec, Z-Val-Val-Arg-NHMec, and Z-Leu-Arg-NHMec, were from Bachem, Bubendorf, Switzerland. The amino acid derivatives were purchased from Novabiochem, Läufelfingen, Switzerland and Bachem, Bubendorf, Switzerland, as well as Fluka, Deisenhofen, Germany. DTT, Brij 35 P, and Triton X-100 were obtained from Fluka; CHAPS was from Sigma, Germany. Diethyl 2-acetamido-2-benzylmalonate was prepared according to reference Citation16. Mathematical data analyses were done with the programs Grafit 4 (Erithacus Software) and GraphPad Prism 4 (GraphPad Software).
Synthesis
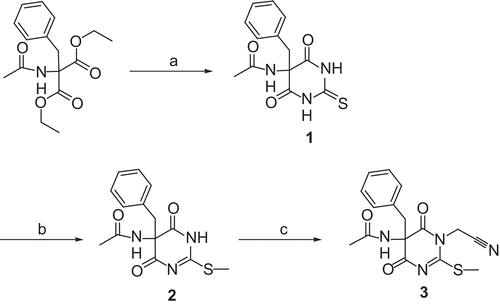
5-Acetamido-5-benzyl-thiobarbituric acid (1)
Diethyl 2-acetamido-2-benzylmalonate (13.11 g, 45 mmol) was added to a solution of sodium (1.04 g, 45 mmol) in dry ethanol (60 mL). After addition of thiourea (3.43 g, 45 mmol), the mixture was refluxed for 37 h. The precipitated sodium salt of the product (8.32 g, 22 mmol) was filtered off, dissolved in water, and treated with 2 N HCl (11 mL) under ice cooling. The precipitate was filtered with suction and dried over P4O10 to obtain sufficiently pure material, yield 52%, mp 283–290°C (dec) (lit.Citation17 288°C (dec) (H2O)); 1H NMR (400 MHz; DMSO-d6): 1.87 (s, 3H, CH3CO), 3.34 (s, 2H, PhCH2C), 6.99–7.04 (m, 2H, 2′-H, 6′-H), 7.24–7.30 (m, 3H, 3′-H, 4′-H, 5′-H), 9.29 (s, 1H, CONHC), 12.18 (s, 2H, 2 × CONHCS); 13C NMR (100 MHz; DMSO-d6): 21.24 (CH3CO), 42.44 (PhCH2C), 63.11 (C-5), 128.11 (C-4′), 128.61, 129.82 (C-3′, C-5′, C-2′, C-6′), 131.63 (C-1′), 168.61, 169.98 (C-4, C-6, CH3CONH), 178.17 (C-2). Anal. found: C, 50.59; H, 4.98; N, 13.28. C13H13N3O3S × H2O requires: C, 50.48; H, 4.89; N, 13.58%.
(±)-2-Methylthio-5-acetamido-5-benzyl-1H,5H-pyrimidine-4,6-dione (2)
Methyl iodide (0.69 mL, 11 mmol) and 1 N NaOH (11 mL) were added to a suspension of 1 (2.90 g, 10 mmol) in acetone (25 mL). After stirring for 5 h at room temperature, the organic solvent was removed in vacuo. The precipitate was filtered off, washed with H2O, and dried. The crude product was recrystallized from acetone/H2O, yield 77 %, mp 285–294°C (dec); 1H NMR (400 MHz; DMSO-d6): 1.85 (s, 3H, CH3CO), 2.18 (s, 3H, CH3S), 3.04 (s, 2H, PhCH2C), 6.97–7.04 (m, 2H, 2′-H, 6′-H), 7.20–7.29 (m, 3H, 3′-H, 4′-H, 5′-H), 9.10 (s, 1H, CH3CONH), 11.98 (s, 1H, 1-H); 13C NMR (100 MHz, DMSO-d6): 13.47 (CH3S), 21.68 (CH3CO), 43.22 (PhCH2C), 65.66 (C-5), 128.07 (C-4′), 128.36, 130.24 (C-3′, C-5′, C-2′, C-6′), 132.41 (C-1′), 169.65, 171.80 (C-4, C-6, CH3CONH), 176.61 (C-2). Anal. found: C, 55.07; H, 4.95; N, 13.76. C14H15N3O3S requires: C, 54.77; H, 5.12; N, 13.35%.
(±)-1-Cyanomethyl-2-methylthio-5-acetamido-5-benzyl-1H,5H-pyrimidine-4,6-dione (3)
A solution of tetrabutylammonium bromide (0.065 g, 0.2 mmol) in 1 N NaOH (2.0 mL) and H2O (4 mL) was added to a solution of 2 (0.6 g, 2.0 mmol) in dichloromethane (6 mL). After addition of bromoacetonitrile (0.15 mL, 2.2 mmol) the mixture was stirred for 12 h at room temperature. Subsequently, the phases were separated and the aqueous phase was extracted with dichloromethane (2 × 20 mL). The combined organic layers were washed with H2O (2 × 15 mL) and brine (15 mL), dried over Na2SO4, and evaporated in vacuo to obtain pure 3, yield 57%, mp 200°C (dec); 1H NMR (500 MHz; CDCl3): 1.88 (s, 3H, CH3CO), 2.26 (s, 3H, CH3S), 3.08 (d, 2J = 12.6 Hz, 1H, PhCHHC), 3.18 (d, 2J = 12.6 Hz, 1H, PhCHHC), 4.82 (s, 2H, NCH2CN), 6.94–6.98 (m, 2H, 2′-H, 6′-H), 7.22–7.30 (m, 3H, 3′-H, 4′-H, 5′-H), 9.41 (br s, 1H, CH3CONH); 13C NMR (125 MHz; CDCl3): 15.34 (SCH3), 21.22 (CH3CO), 31.25 (NCH2CN), 42.89 (PhCH2C), 65.88 (C-5), 114.12 (NCH2CN), 128.05 (C-4′), 128.41, 129.74 (C-3′, C-5′, C-2′, C-6′), 131.56 (C-1′), 169.84, 170.00, 170.20 (C-4, C-6, CH3CONH), 173.77 (C-2); MS (EI) m/z (%): 344 (3, M+.), 189 (100, RDA: [dienophile]+.), 156 (57, RDA: [diene + H]+), 147 (18, [dienophile-C2H2O]+.), 91 (42, [C7H7]+). Anal. found: C, 53.18; H, 5.20; N, 15.11; C16H16N4O3S requires: C, 55.80; H, 4.68; N, 16.27%.
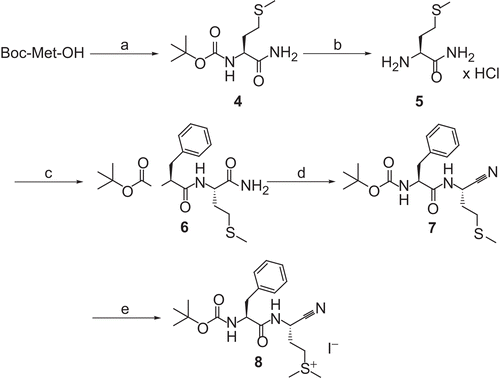
N-(tert-Butoxycarbonyl)-methionine amide (4)
General procedure for amides 4, 6, and 9 Boc-Met-OH (4.00 g, 16 mmol) was dissolved in THF (15 mL) and cooled to −25°C. Subsequently, N-methylmorpholine (4.87 mL, 16 mmol) and isobutyl chloroformate (2.11 mL, 16 mmol) were sequentially added under vigorous stirring. Immediately after the formation of the white precipitate of N-methylmorpholine hydrochloride, 25% NH3 (6 mL, 80 mmol) was added. The resulting mixture was allowed to warm to room temperature. After 2 h, THF was removed under reduced pressure and the residual aqueous mixture was diluted with a small volume of H2O, adjusted to pH 1 (10% NaHSO4), and extracted with ethyl acetate (3 × 50 mL). The combined organic layers were washed with H2O (20 mL), sat. NaHCO3 (2 × 20 mL), and brine (20 mL), dried over Na2SO4 and evaporated to dryness to obtain pure 4, yield 86%, mp 122–123°C (lit.Citation18 118–119°C); 1H NMR (500 MHz; DMSO-d6): 1.37 (s, 9H, ((CH3)3C), 1.69–1.78 (m, 1H, SCHHCH2), 1.80–1.89 (m, 1H, SCHHCH2), 2.02 (s, 3H, CH3S), 2.36–2.47 (m, 2H, CH2CH2CH), 3.89–3.96 (m, 1H, NHCHCO), 6.80 (d, 3J = 8.2 Hz, 1H, NHCHCO), 6.94 (s, 1H, CONHH), 7.20 (s, 1H, CONHH); 13C NMR (125 MHz; DMSO-d6): 14.76 (CH3S), 28.32 ((CH3)3C), 29.95 (SCH2CH2), 31.87 (CH2CH2CH), 53.53 (NHCHCO), 78.15 ((CH3)3CO), 155.50 (OCONH), 173.87 (CHCONH2). Anal. found: C, 46.41; H, 8.42; N, 10.94; C10H20N2O3S × 1/2 H2O requires: C, 46.67; H, 8.22; N, 10.89%.
Methionine amide hydrochloride (5)
Compound 4 (1.0 g, 4.0 mmol) was dissolved in 4 M HCl/dioxane (20 mL) under an atmosphere of argon. After a few minutes the product precipitated. The mixture was kept at room temperature for 1 h. The solvent was removed in vacuo, and the residue suspended in petroleum ether, filtered off, and dried in a desiccator, yield 97%, mp 221°C (dec) (lit.Citation19 mp 189–190°C); 1H NMR (500 MHz; DMSO-d6): 1.98–2.04 (m, 2H, SCH2CH2), 2.05 (s, 3H, CH3S), 2.49–2.54 (m, 2H, CH2CH2CH), 3.79 (t, 3J = 6.1 Hz, 1H, H3N+CHCO), 7.51 (s, 1H, CONHH), 8.02 (s, 1H, CONHH), 8.35 (br s, 3H, H3N+CHCO); 13C NMR (125 MHz; DMSO-d6): δ 14.61 (CH3S), 28.49 (SCH2CH2), 30.88 (CH2CH2CH), 51.68 (H3N+CHCO), 170.03 (CHCONH2); Anal. found: C, 31.87; H, 8.16; N, 15.49; C5H12N2OS × HCl requires: C, 32.52; H, 7.09; N: 15.17%.
N-(tert-Butoxycarbonyl)-phenylalanyl-methionine amide (6)
Preparation was done according to the general procedure given for compound 4 using 0.93 g Boc-Phe-OH (3.5 mmol). Instead of NH3, a solution of 5 (0.7 g, 3.8 mmol) in H2O (0.5 mL) and 1 N NaOH (3.8 mL) was used, yield 99%, mp 187°C; 1H NMR (500 MHz; DMSO-d6): 1.30 (s, 9H, (CH3)3C), 1.72–1.82 (m, 1H, SCHHCH2), 1.90–1.99 (m, 1H; SCHHCH2), 2.02 (s, 3H, CH3S), 2.33–2.47 (m, 2H, CH2CH2CH), 2.75 (dd, 2J = 13.6 Hz, 3J = 10.1 Hz, 1H, PhCHHCH), 2.97 (dd, 2J = 13.8 Hz, 3J = 4.6 Hz, 1H, PhCHHCH), 4.11–4.18 (m, 1H), 4.25–4.35 (m, 1H) (NHCHCO-Met, NHCHCO-Phe); 6.99 (d, 3J = 8.4 Hz, 1H, NHCHCO-Phe), 7.06 (s, 1H, CONHH), 7.15–7.21 (m, 2H, 2′-H, 6′-H), 7.22–7.28 (m, 4H, 3′-H, 4′-H, 5′-H, CONHH), 7.89 (d, 3J = 8.2 Hz, 1H, NHCHCO-Met); 13C NMR (125 MHz; DMSO-d6): 14.76 (SCH3), 28.25 ((CH3)3C), 29.58 (SCH2CH2), 32.18 (CH2CH2CH), 37.15 (PhCH2CH), 51.75, 56.04 (NHCHCO-Met, NHCHCO-Phe), 78.30 ((CH3)3CO), 126.31 (C-4′), 128.15 (C-3′, C-5′), 129.32 (C-2′, C-6′), 138.26 (C-1′), 155.45 (OCONH), 171.66, 172.98 (CHCONH, CONH2). Anal. found: C, 54.34; H, 9.24; N, 11.94; C19H29N3O4S × H2O requires: C, 55.13; H, 7.56; N, 10.16%.
N-(tert-Butoxycarbonyl)-phenylalanyl-methionine nitrile (7)
To a solution of 6 (0.80 g, 2.0 mmol) in DMF (5 mL), cyanuric chloride (0.30 g, 2.0 mmol) was added as a solid under ice cooling. After stirring for 2 h at room temperature, ice-cold sat. NaHCO3 (10 mL) was added and the resulting mixture was extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with H2O (3 × 10 mL) and brine (10 mL), dried over Na2SO4, and evaporated in vacuo. The obtained residue was recrystallized from n-hexane/ethyl acetate, yield 60%, mp 127–128°C; 1H NMR (500 MHz; CDCl3): 1.40 (s, 9H, (CH3)3C), 1.96–2.03 (m, 2H, SCH2CH2), 2.05 (s, 3H, CH3S), 2.49–2.57 (m, 2H, CH2CH2CH), 3.03 (br s, 1H, PhCHHCH), 3.04 (br s, 1H, PhCHHCH), 4.25–4.31 (m, 1H, NHCHCO), 4.93–5.01 (m, 1H, NHCHCN), 5.04 (br s, 1H, NHCHCO), 6.72 (d, 3J = 8.2 Hz, 1H, NHCHCN), 7.15–7.19 (m, 2H, 2′-H, 6′-H); 7.22–7.27 (m, 1H, 4′-H); 7.28–7.33 (m, 2H, 3′-H, 5′-H); 13C NMR (125 MHz; CDCl3): 15.34 (CH3S), 28.23 ((CH3)3C), 29.50 (SCH2CH2), 31.67 (NHCHCN), 38.00, 39.50 (PhCH2CH, CH2CH2CH), 55.79 (NHCHCO), 80.72 ((CH3)3CO), 117.54 (NHCHCN), 127.25 (C-4′), 128.89, 129.25 (C-3′, C-5′, C2′, C-6′), 136.00 (C-1′), 155.54 (OCONH), 171.06 (CHCONH); MS (ESI) m/z (%) (pos): 416 (7, [M + K]+), 400 (11, [M + Na]+), 395 (67, [M + NH4]+), 378 (83, [M + H]+), 322 (100, [M-(CH3)2CCH2 + H]+), 278 (94, [M-(CH3)2CCH2-CO2 + H]+) (neg): 436 (7, [M + CH3COO]−), 376 (100, [M – H]−), 302 (72, [M-tBuOH-H]−). Anal. found: C, 59.96; H, 7.66; N; 10.95; C19H27N3O3S requires: C, 60.45; H, 7.21; N, 11.13%.
N-(tert-Butoxycarbonyl)-phenylalanyl-S-methylmethionine nitrile iodide (8)
Methyl iodide (0.16 mL, 2.60 mmol) was added to a solution of 7 (0.10 g, 0.26 mmol) in ethyl acetate (1 mL). The mixture was stirred for 3 days at room temperature under the exclusion of light. The residue obtained after concentration in vacuo was treated with dry ether. The precipitate was filtered off and washed carefully with ether, yield 36%; 1H NMR (500 MHz; DMSO-d6): 1.31 (s, 9H, (CH3)3C), 2.19–2.37 (m, 2H, S+CH2CH2), 2.81 (dd, 2J = 13.6 Hz, 3J = 9.5 Hz, 1H, PhCHHCH), 2.88 (s, 3H, CH3CH3S+), 2.91 (s, 3H, CH3CH3S+), 2.95 (dd, 2J = 13.7 Hz, 3J = 5.2 Hz, 1H, PhCHHCH), 3.20–3.35 (m, 2H, CH2CH2CH), 4.05–4.13 (m, 1H, NHCHCO), 4.94–5.02 (m, 1H, NHCHCN), 7.13 (d, 3J = 7.6 Hz, NHCHCO), 7.16–7.30 (m, 5H, Ph-H), 8.88 (d, 3J = 7.9 Hz, 1H, NHCHCN); 13C NMR (125 MHz, DMSO-d6): 24.72 (CH3CH3S+), 24.75 (CH3CH3S+), 26.55 (NHCHCN), 28.28 ((CH3)3C), 37.02, 38.33, 38.69 (S+CH2CH2, PhCH2CH, CH2CH2CH), 56.03 (NHCHCO), 78.44 ((CH3)3CO), 118.41 (NHCHCN), 126.52 (C-4′), 128.26 (C-3′, C-5′), 129.29 (C-2′, C-6′), 137.70 (C-1′), 155.51 (OCONH), 172.28 (CHCONH); MS (ESI) m/z (%) (pos): 392 (100, C20H30N3O3S+) (neg): 127 (100, I−).
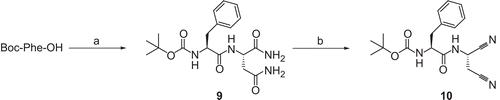
N-(tert-Butoxycarbonyl)-phenylalanyl-asparagine amide (9)
Preparation was done according to the general procedure given for compound 4 using 0.80 g Boc-Phe-OH (3.0 mmol). Instead of NH3, a solution of H-Asn-NH2 × HCl (0.50 g, 3.0 mmol) in H2O (0.5 mL) and 1 N NaOH (3.0 mL) was used. Differing from the general procedure, the precipitate obtained after the removal of THF was treated with H2O (5 mL) and the suspension acidified to pH 1 (10% NaHSO4). The solid was filtered off and washed with H2O (2 × 15 mL), sat. NaHCO3 (15 mL), and H2O (15 mL) and dried in a desiccator over P4O10, yield 79%, mp 179–181°C (dec); 1H NMR (500 MHz; DMSO-d6): 1.30 (s, 9H, (CH3)3C), 2.45 (dd, 2J = 15.5 Hz, 3J = 6.1 Hz, 1H, H2NCOCHHCH), 2.51 (dd, 2J = 15.8 Hz, 3J = 6.1 Hz, 1H, H2NCOCHHCH), 2.73 (dd, 2J = 13.7 Hz, 3J = 10.5 Hz, 1H, PhCHHCH), 2.98 (dd, 2J = 14.0 Hz, 3J = 4.2 Hz, PhCHHCH), 4.07–4.15 (m, 1H), 4.38–4.53 (m, 1H) (2 × NHCHCO), 6.86 (s, 1H, CONHH), 6.97 (s, 1H, CONHH), 7.03 (d, 3J = 8.2 Hz, 1H, NHCHCO-Phe), 7.05 (s, 1H, CONHH), 7.13–7.30 (m, 5H, Ph-H), 7.33 (s, 1H, CONHH), 8.11 (d, 3J = 7.9 Hz, 1H, NHCHCO-Asn); 13C NMR (125 MHz; DMSO-d6): 28.27 ((CH3)C), 36.87, 37.15 (COCH2CH-Asn, PhCH2CH-Phe), 49.65, 56.25 (NHCHCO-Asn, NHCHCO-Phe), 78.48 ((CH3)3CO), 126.30 (C-4′), 128.16, 129.32 (C-3′, C-5′, C-2′, C-6′), 138.29 (C-1′), 155.61 (OCONH), 171.45, 171.96, 172.87 (CONH, CHCONH2, CH2CONH2). Anal. found: C, 52.73; H, 7.24; N, 13.11; C18H26N4O5 × 2 H2O requires: C, 52.16; H, 7.30; N, 13.52%.
N-(tert-Butoxycarbonyl)-phenylalanyl-cyanoalanine nitrile (10)
To a solution of 9 (0.80 g, 2.1 mmol) in DMF (10 mL), cyanuric chloride (0.62 g, 4.2 mmol) was added as a solid under ice cooling. After stirring for 2 h at room temperature, ice-cold sat. NaHCO3 (10 mL) was added and the resulting mixture was extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with H2O (3 × 10 mL) and brine (10 mL), dried over Na2SO4 and evaporated in vacuo, yield 99%, mp 117–119°C, [α]D20 = −8.53 (c = 1.17, MeOH); 1H NMR (500 MHz; CDCl3): 1.39 (s, 9H, (CH3)3C), 2.89 (d, 3J = 6.1 Hz, 2H, CHCH2CN), 2.97 (dd, 2J = 13.8 Hz, 3J = 7.8 Hz, 1H, PhCHHCH), 3.02–3.11 (m, 1H, PhCHHCH), 4.32–4.43 (m, 1H, NHCHCO), 5.05–5.11 (m, 1H, NHCHCN), 5.23 (d, 3J = 6.3 Hz, 1H, NHCHCO), 7.14 (d, 3J = 6.9 Hz, 2H, 2′-H, 6′-H), 7.21–7.26 (m, 1H, 4′-H), 7.27–7.31 (m, 2H, 3′-H, 5′-H), 7.74 (br s, 1H, NHCHCN); 13C NMR (125 MHz; CDCl3): 22.63 (CHCH2CN), 28.26 ((CH3)3C), 37.22, 37.95 (NHCHCN, PhCH2CH), 55.71 (NHCHCO), 81.00 ((CH3)3CO), 114.55, 115.39 (CH2CN, CHCN), 127.29 (C-4′), 128.83, 129.16 (C-3′, C-5′, C-2′, C-6′), 135.84 (C-1′), 155.75 (OCONH), 171.93 (CHCONH); MS (ESI) m/z (%) (pos): 381 (8, [M + K]+), 365 (15, [M + Na]+), 360 (62, [M + NH4]+), 343 (22, [M + H]+), 287 (100, [M-(CH3)2CCH2 + H]+), 243 (75, [M-(CH3)2CCH2-CO2 + H]+) (neg): 341 (100, [M – H]−), 267 (64, [M-tBuOH – H]−). Anal. found: C, 62.22; H, 6.96; N, 16.08; C18H22N4O3 requires: C, 63.14; H, 6.48; N, 16.36%.
Inhibition experiments
Papain
Enzyme activities were determined by spectrophotometric detection of the product p-nitroaniline at 25°C in a multi-cell holder (parallel measurements for each single inhibitor determination; final volume 1 mL) at a wavelength of 405 nm. A 4 mM stock solution of the chromogenic substrate Z-Phe-Arg-NHNp was prepared in DMSO; the final concentration was 200 μM (= 0.207Km)Citation20. The assay medium was 0.1 M sodium phosphate pH 6.5, 2.5 mM EDTA, 300 μM DTT, and 12% DMSO. Stock solutions of the inhibitors were prepared in DMSO. In the absence of inhibitor, 70 μL of DMSO was added to the cuvette. A papain stock solution was prepared in 1 mM HCl. For daily activation, the papain stock solution was diluted 1:100 in 0.1 M sodium phosphate pH 6.5, 2.5 mM EDTA, and 15 mM DTT and incubated at 25°C for 1 h. The activated enzyme was kept on ice. After thermal equilibration, the reaction was initiated by addition of the enzyme (20 μL); its final concentration catalyzed the conversion of the substrate at a rate of 1–2 μM/min. Progress curves were monitored over 10 min. Rates were determined for eight different inhibitor concentrations including the control in duplicate.
Cathepsins
Enzyme activities were calculated from kinetic measurements performed by fluorimetric detection of the product 7-amino-4-methylcoumarin at 37°C in a stirred cuvette. The wavelengths for excitation and emission were 360 nm and 440 nm, respectively. The reaction volume of the assay was 2 mL. To assay cathepsin L, Z-Phe-Arg-NHMec was used as substrate at a concentration of 10 μM (= 1.89Km)Citation20 in 100 mM sodium phosphate pH 6.0, 100 mM NaCl, 5 mM EDTA, 0.01% Brij 35, 25 μM DTT, and 1% DMSO. For cathepsin S, Z-Val-Val-Arg-NHMec was chosen as substrate at a concentration of 40 μM (= 2.08 Km)Citation20 in 50 mM potassium phosphate pH 6.5, 50 mM NaCl, 2 mM EDTA, 0.01% Triton X-100, 25 μM DTT, and 1% DMSO. In the cathepsin K assay, Z-Leu-Arg-NHMec was used as substrate at a concentration of 20 μM (= 3.23Km)Citation20 in 100 mM sodium citrate pH 5.0, 100 mM NaCl, 1 mM EDTA, 0.01% CHAPS, 25 μM DTT, and 1% DMSO. Stock solutions of the substrates were prepared in DMSO in a 1000-fold higher concentration than the final concentrations. Dilutions were done in the corresponding assay medium without DTT and DMSO. Stock solutions of the inhibitors and dilutions were prepared in DMSO. In the absence of inhibitor, 20 μL of DMSO was added to the cuvette. The enzyme dilutions were daily prepared from a stock with the corresponding assay medium without DMSO, containing 5 mM DTT (the complete amount of DTT required for the final concentration noted above), and kept on ice. After thermal equilibration, 10 μL of the enzyme solution was added and product formation was monitored over 5 min. The inhibition constants were obtained from measurements at 10–12 different inhibitor concentrations and duplicate or triplicate measurements in the absence of the inhibitor.
Determination of kinetic parameters for the inhibition of cysteine proteases
The progress curves of the cysteine protease-catalyzed reactions in the presence of the dipeptide nitriles 7, 8, and 10 were nearly linear, with a slight deviation from linearity—also in the reactions without inhibitor—in the case of cathepsin S. The apparent inhibition constant Ki’ was determined by fitting:
to the experimental data, where v is the rate, v0 is the rate in absence of inhibitor, and [I] is the inhibitor concentrationCitation21. The true inhibition constant Ki was calculated by correction of Ki9 according to:
where [S] is the substrate concentration and Km is the Michaelis constantCitation22.
Structure calculations
The structure of the l-phenylalanine-derived enantiomer of 3 was calculated in the program Sybyl 7.3 using the Tripos force field. A simulated annealing was performed by heating to 700 K for 1000 fs followed by cooling to 200 K within 1000 fs. Low-energy conformers were selected and their structure subsequently energetically minimized using the Powell gradient method (0.05 kcal/mol, 1000 iterations). A low-energy conformation with values of −69° and +128° for ϕ and ψ, respectively, was selected and subjected to molecular dynamics simulation at different temperatures of 100 K, 300 K, 600 K, and 800 K. The chosen parameters were as follows: ensemble: NTV (constant temperature and volume); initial velocity: Boltzmann; time length: 1000 fs; coupling: 100 fs; step: 1 s; number of maximum iterations: 100. Each simulation was followed by energy minimization as described above. After each dynamics/minimization run the dihedral angles were measured, yielding average values for ϕ and ψ of –(80 ± 10)° and +(124 ± 3)°, respectively.
The structure of the corresponding open-chain N-acetyl-phenylalanyl-glycine nitrile was generated by constraining the dihedral angles to ϕ = −139° and ψ = +135°, typical values for the extended β-strand conformationCitation23, and energy minimization as described above.
Results and discussion
Synthesis
A heterocyclic mimetic of N-acetyl-phenylalanyl-glycine nitrile was designed by connecting the α-carbon of phenylalanine with the peptide bond nitrogen by a -CO−N=C(SCH3)- chain (). To afford this heterocyclic dipeptide derivative 3, the synthesis started with the preparation of the thiobarbituric acid 1 by cyclocondensation of thiourea and diethyl 2-acetamido-2-benzylmalonate. This intermediate was S-methylated to obtain 2 in good yield. Compound 2 was converted to the final product 3 by N-alkylation with bromoacetonitrile under phase transfer catalysis. The chemoselective alkylation at the N-atom was confirmed by NMR as well as by EI-MS due to the occurrence of the retro-Diels-Alder fragmentation of the molecular ion of 3 (see “Materials and methods”).
Scheme 1. Synthesis of compound 3. Reagents and conditions: (a) (NH2)2CS, NaOEt, EtOH, reflux; (b) 1 N NaOH, CH3I, acetone, room temperature; (c) BrCH2CN, (nBu)4NBr, 1 N NaOH, H2O/CH2Cl2.
The synthesis of the open-chain dipeptide nitrile 8 containing a methionine-derived dimethylsulfonium moiety is outlined in . The route involved the formation of the Boc-protected methionine amide 4 and deprotection to the amino acid amide 5. Coupling of 5 with Boc-Phe-OH and treatment of the resulting dipeptide amide 6 with cyanuric chloride in DMFCitation24 gave the dipeptide nitrile 7. The dimethylsulfonium derivative 8 was finally obtained by S-methylation of 7 with methyl iodide in moderate yield.
Scheme 2. Synthesis of compound 8. Reagents and conditions: (a) (i) N- methylmorpholine, ClCO2CH2CH(CH3)2, THF; (ii) 25% NH3, −25°C to room temperature; (b) 4 N HCl/dioxane, room temperature; (c) (i) Boc-Phe-OH, N-methylmorpholine, ClCO2CH2CH(CH3)2, THF; (ii) 7 in 1 N NaOH/H2O, −25°C to room temperature; (d) (ClCN)3, DMF, room temperature; (e) CH3I, ethyl acetate, room temperature.
The dipeptide-derived dinitrile 10 () was prepared by the same methodology as that applied for the preparation of 8. Dehydratization of the two primary amide groups in 4 with cyanuric chloride in DMF afforded 10 in very good yield.
Scheme 3. Synthesis of compound 10. Reagents and conditions: (a) (i) N-methylmorpholine, ClCO2CH2CH(CH3)2, THF; (ii) H-Asn-NH2 × HCl, H2O, 1 N NaOH, −25°C to room temperature; (b) (ClCN)3, DMF, room temperature.
Inhibitory activity
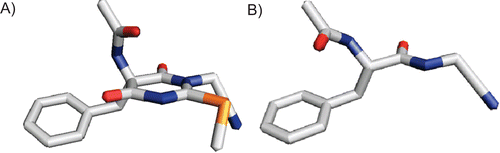
The heterocyclic nitrile 3 was prepared to combine the dipeptide nitrile pharmacophore and the structural requirements for a thioimidate trapping, i.e. the cyanomethyl group and a neighboring methylthio group attached to an sp2 hybridized carbon. Despite its dipeptide nitrile structure, compound 3 did not show any inhibitory activity against papain. This can be explained by different reasons. The first is that the amide bond between the P2 and P1 positions lacks a proton for hydrogen bonding, and the second is that the “nonpeptidic” parts of the pyrimidine ring may lead to steric hindrance in the active site of the enzyme. Another explanation for the lack of inhibitory activity concerns the conformational rigidity of the compound. It is widely accepted that proteases generally recognize their substrates and inhibitors in an extended β-strand conformationCitation25. The conformation of the main chain of a peptide is described by the dihedral angles ϕ and ψ (). The pyrimidine ring in compound 3 restricts the possible values of the dihedral angles. Simulated annealingCitation26 and molecular dynamics simulationsCitation27 for the l-phenylanine-derived enantiomer of 3 suggested for ϕ and ψ average values of −80° and +124°, respectively. This matches approximately the I + 1 position of a β-II turn (ϕi+ 1 = −60°, ψi+ 1 = +120°)Citation28. Therefore, one can conclude that the peptidic backbone in compound 3 is restricted in a rather bended conformation, which cannot be recognized by the proteases (). A decrease in the cysteine protease-inhibiting activity as a result of rigidization was also observed with similarly constrained aldehyde and vinyl sulfone inhibitorsCitation30. However, the fact that the dihedral angles meet those of the β-II turn makes this type of compound interesting for the design of turn mimetics.
Figure 2. The dihedral angles ϕ and ψ at the peptidic backbone. ϕ is defined by the atoms Ci – 1, Ni, Cαi, and Ci and ψ by Ni, Cαi, Ci, and Ni + 1.
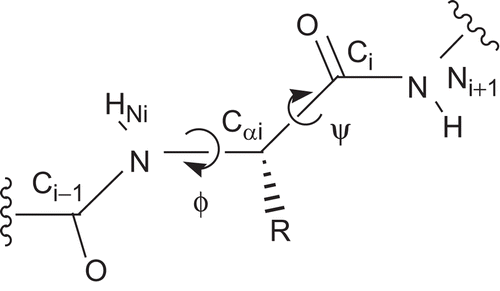
Figure 3. (A) Three-dimensional representation of the l-phenylalanine-derived enantiomer of compound 3. The values for the dihedral angles are ϕ = −80° and ψ = +124°. (B) For comparison, the corresponding open-chain N-acetyl-phenylalanyl-glycine nitrile in the extended, bioactive conformation (ϕ = −139°, ψ = +135°) is shown. Images prepared with PyMOLCitation29.
To enable the inhibitors to adopt the bioactive conformation, open-chain dipeptide nitriles containing the electrophilic center in the side chain of the P1 position were designed. This was promising, as the P1 position in papain-like cysteine proteases is mostly solvent exposed, and therefore a broad range of functionalities should be tolerated thereCitation31. The electrophiles considered were a second cyano group in the 1,2-position as realized in the cyanoalanine nitrile 10 and the γ-carbon attached to a positively charged sulfur atom in the methionine-derived sulfonium salt 8. The cyanoalanine nitrile was selected as dielectrophile because it has been demonstrated that succinic dinitrile reacts with mercaptoacetic acid to form the corresponding 2-alkylthio-5-aminopyrrole, according to Citation32. In the methionine-derived sulfonium-salt 8, the nitrogen of the cyano group could attack the γ-carbon in an SN2 reaction after formation of the thioimidate, with the active-site thiol leading to an enzyme-bound five-membered cyclic thioimidate. According to Baldwin’s rules on ring closing reactions, this represents a 5-exo-tet process, which—as all exo-tet ring closures—is favoredCitation33. Methionine-derived sulfonium salts have been successfully applied to the synthesis of pyrrolinonesCitation34.
Figure 4. Formation of 2-alkylthio-5-aminopyrroles from succinic dinitrile and thioles (R = CH2COOH) according to Elnagdi et al.Citation32.
The three dipeptide nitriles 7, 8, and 10 were evaluated against the cysteine proteases papain and cathepsin L, S, and K (). As expected, derivative 7, containing no electrophilic group in the P1 side chain, showed a reasonable inhibition against all enzymes in the higher nanomolar to the lower micromolar range. This confirms that the Phe moiety in P2, the N-terminal Boc-group as well as the Met residue, as one among a number of possible residues in P1, can be well adopted by all considered enzymes. Typical for peptide-derived nitriles, its inhibition behavior was time-independentCitation11,Citation20.
Table 1. Inhibition of cathepsin L, cathepsin S, cathepsin K, and papain by dipeptide nitriles with electrophilic side chains in the P1 position.
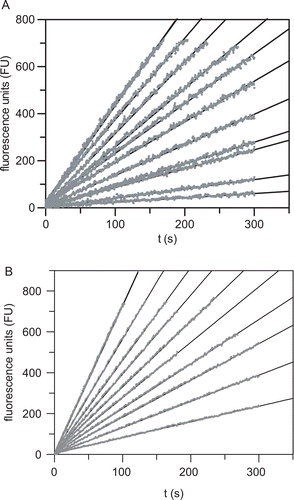
Compounds 8 and 10 were also potent inhibitors of cysteine proteases, with Ki values of similar magnitude (). However, as the dipeptide nitriles 8 and 10 were designed as mechanism-based inactivators, a two-step mechanism for their interactions with the enzymes was expected. The first step should comprise the reversible formation of the thioimidate by reaction of the active-site thiol with the cyano group, followed by the second, irreversible intramolecular reaction of the thioimidate nitrogen with the carbon-electrophile in the P1 side chain. However, the evaluation of the two side chain-modified dipeptides revealed linear progress curves for each inhibitor concentration (). The sulfonium salt 8 showed similar inhibition constants when compared to its nonmethylated counterpart 7. In contrast, the inhibitory activity of the dinitrile 10 was significantly weaker than that of 7, especially for the cathepsins L and K, as well as papain. This is probably due to the fact that the cyanomethyl residue in P1 is shorter than the Met-derived side chains of the derivatives 7 and 8 in the according position, and thus the interactions to the P1 or possibly the P19 binding sites are less attractive than in the case of 10. The linearity of the substrate conversion observed in the inhibition experiments for the compounds 8 and 10 indicates that no covalent trapping of the thioimidate adduct occurred, as irreversible inhibition is generally associated with a slow two-step mechanism leading to steady-state rates equaling zeroCitation35,Citation36. Therefore, it can be concluded that a further covalent modification of the thioimidate nitrogen by a nearby electrophile cannot be achieved in the nitrile-derived enzyme-inhibitor complexes.
Figure 5. (A) Cathepsin S-catalyzed hydrolysis of Z-Val-Val-Arg-NHMec (40 μM) in the presence of increasing concentrations of compound 8 (from top to bottom: 0, 0, 0.3, 0.5, 1.0, 2.0, 3.0, 5.0, 7.0, 10, 20, 100 μM) in 50 mM potassium phosphate pH 6.5, 50 mM NaCl, 2 mM EDTA, 0.01% Triton X-100, 25 μM DTT, 1% DMSO, 37°C. (B) Cathepsin L-catalyzed hydrolysis of Z-Phe-Arg-NHMec (10 μM) in the presence of increasing concentrations of compound 10 (from top to bottom: 0, 0, 1, 2, 3, 4, 5, 7, 10, 15, 50 μM) in 0.1 M sodium phosphate pH 6.0, 0.1 M NaCl, 5 mM EDTA, 0.01% Brij 35, 25 μM DTT, 1% DMSO, 37°C. The reactions were initiated by addition of the enzyme. Fluorescence emission at 440 nm was measured after excitation at 360 nm. Fluorescence units (FU) were corrected for background fluorescence.
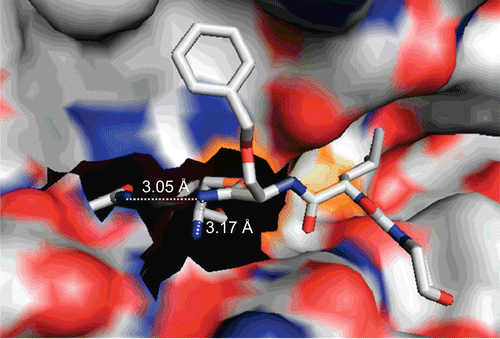
This might be because the thioimidate adduct—despite the fact that it is planar—is stabilized by hydrogen bond contacts between the nitrogen and the NH of the side chain of Gln 19, similar to the stabilizing interaction between the tetrahedral oxyanion intermediate and the oxyanion hole formed by the backbone NH of Cys 25 and the side chain amide group of Gln 19. Indeed, such a stabilization of the enzyme-bound thioimidate becomes evident from a published crystal structure of cathepsin S complexed with a dipeptide nitrile, morpholinocarbonyl-leucyl-O-benzylserine nitrileCitation37. The nitrogen atom of the thioimidate group formed from the cyano group by attack of the active-site thiol is in hydrogen bond distances of 3.17 Å and 3.05 Å to the backbone nitrogen of Cys 25 and the side chain nitrogen of Gln 19, respectively (). Such information from protein crystallography and our kinetic results indicate the limitations of an approach to irreversibly inhibit cysteine proteases by applying thioimidate-trapping cascade reactions.
Figure 6. Crystal structure of the complex between cathepsin S and morpholinocarbonyl-leucyl-O-benzylserine nitrile. The thioimidate nitrogen is in hydrogen bond distance to the side chain nitrogen of Gln 19 and the main chain nitrogen of Cys 25. PDB entry 1MS6Citation37, image prepared with PyMOLCitation29.
Acknowledgments
We are grateful to Dr. Huy Hoang, Brisbane, for his help in the structural calculations and valuable discussions. The advice of Dr. Klaus Schilling, Jena, in the assay of the cathepsins is cordially acknowledged.
Declaration of interest: The authors report no conflicts of interest.
Related Research Data
References
- Lecaille F, Kaleta J, Brömme D. Chem Rev 2002;102:4459–88.
- Powers JC, Asgian JL, Ekici öD, James KE. Chem Rev 2002;102:4639–750.
- Vicik R, Busemann M, Baumann K, Schirmeister T. Curr Top Med Chem 2006;6:331–53.
- Schaschke N, Assfalg-Machleidt I, Machleidt W, Turk D, Moroder L. Bioorg Med Chem 1997;5:1789–97.
- Schirmeister T. J Med Chem 1999;42:560–72.
- Shaw E. Methods Enzymol 1994;244:649–56.
- Brömme D, Klaus JL, Okamoto K, Rasnick D, Palmer JT. Biochem J 1996;315:85–9.
- Darkins P, Gilmore BF, Hawthorne SJ, Healy A, Moncrieff H, McCarthy N, McKervey MA, Brömme D, Pagano M, Walker B. Chem Biol Drug Des 2007;69:170–9.
- Santos MMM, Moreira R. Mini-Rev Med Chem 2007;7:1040–50.
- Moon JB, Coleman RS, Hanzlik RP. J Am Chem Soc 1986;108:1350–1.
- Löser R, Frizler M, Schilling K, Gütschow M. Angew Chem Int Ed Engl 2008;47:4331–4.
- Wright SW, Ammirati MJ, Andrews KM, Brodeur AM, Danley DE, Doran SD, Lillquist JS, McClure LD, McPherson RK, Orena SJ, Parker JC, Polivkova J, Qiu X, Soeller WC, Soglia CB, Treadway JL, VanVolkenburg MA, Wang H, Wilder DC, Olson TV. J Med Chem 2006;49:3068–76.
- Gütschow M, Powers JC. J Org Chem 2001;66:4723–7.
- Tietze LF. Chem Rev 1996;96:115–36.
- Linnevers CJ, McGrath ME, Armstrong R, Mistry FR, Barnes MG, Klaus JL, Palmer JT, Katz BA, Brömme D. Protein Sci 1997;6:919–21.
- Snyder HR, Sekleton JF, Lewis CD. J Am Chem Soc 1945;67:310–12.
- Singh P, Berlinguet L. Can J Chem 1964;42:605–9.
- Boomgaarden M, Heinrich G, Henklein P. Pharmazie 1992;47:710–11.
- Johnson HE, Crosby DG. J Org Chem 1962;27:798–802.
- Löser R, Schilling K, Dimmig E, Gütschow M. J Med Chem 2005;48:7688–707.
- Copeland RA. Enzymes: A Practical Indroduction to Structure, Mechanism and Data Analysis. New York: Wiley-VCH, 2005:282–3.
- Cheng YC, Prusoff WH. Biochem Pharmacol 1973;22:3099–108.
- Loughlin WA, Tyndall JDA, Glenn MP, Fairlie DP. Chem Rev 2004;104:6085–117.
- Olah GA, Narang SC, Fung AP, Gupta BGB. Synthesis 1980:657–8.
- Tyndall JDA, Nall T, Fairlie DP. Chem Rev 2005;105:973–99.
- Wilson SR, Cui W. Biopolymers 1990;29:225–35.
- van Gunsteren W, Berendsen HJC. Angew Chem Int Ed Engl 1990;29:992–1023.
- Sewald N, Jakubke HD.Peptides: Chemistry and Biology. Weinheim: Wiley-VCH, 2002:40.
- DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific, 2005.
- Scheidt KA, Roush WA, McKerrow JH, Selzer PM, Hansell E, Rosenthal PJ. Bioorg Med Chem 1998;6:2477–94.
- Turk D, Guncar G, Podobnik M, Turk B. Biol Chem 1998;379:137–47.
- Elnagdi MH, Khalifa MAE, Ibraheim MKA, Elmoghayar MRH. J Heterocyclic Chem 1981;18:877–9.
- Clayden J, Greeves N, Warren S, Wothers P. Organic Chemistry. Oxford: Oxford University Press, 2001:1140–3.
- Freidinger RM, Perlow DS, Veber DF. J Org Chem 1982;47:104–9.
- Tian WX, Tsou CL. Biochemistry 1982;21:1028–32.
- Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery. Hoboken: Wiley-Interscience, 2005:215–19.
- Ward YD, Thomson DS, Frye LL, Cywin CL, Morwick T, Emmanuel MJ, Zindell R, McNeil D, Bekkali Y, Girardot M, Hrapchak M, DeTuri M, Crane K, White D, Pav S, Wang Y, Hao M-H, Grygon CA, Labadia ME, Freeman DM, Davidson W, Hopkins JL, Brown ML, Spero DM. J Med Chem 2002;45:5471–82.