Abstract
Carnitine acetyltransferase (CrAT; EC 2.3.1.7) catalyzes the reversible transfer of acetyl groups between acetyl-coenzyme A (acetyl-CoA) and L-carnitine; it also regulates the cellular pool of CoA and the availability of activated acetyl groups. In this study, biochemical measurements, saturation transfer difference (STD) nuclear magnetic resonance (NMR) spectroscopy, and molecular docking were applied to give insights into the CrAT binding of a synthetic inhibitor, the cardioprotective drug mildronate (3-(2,2,2-trimethylhydrazinium)-propionate). The obtained results show that mildronate inhibits CrAT in a competitive manner through binding to the carnitine binding site, not the acetyl-CoA binding site. The bound conformation of mildronate closely resembles that of carnitine except for the orientation of the trimethylammonium group, which in the mildronate molecule is exposed to the solvent. The dissociation constant of the mildronate CrAT complex is approximately 0.1 mM, and the Ki is 1.6 mM. The results suggest that the cardioprotective effect of mildronate might be partially mediated by CrAT inhibition and concomitant regulation of cellular energy metabolism pathways.
Introduction
Carnitine acetyltransferase (CrAT; EC 2.3.1.7) catalyzes the reversible transfer of acetyl groups between acetyl-coenzyme A (acetyl-CoA) and L-carnitine () in mammalian mitochondrial matrix, endoplasmic reticulum lumen, and peroxisomesCitation1. Since CrAT is homologous to other carnitine acyltransferases and, particularly, carnitine palmitoyltransferase 1 (CPT I; EC 2.3.1.21), which regulates long chain fatty acid metabolism, studies of the enzyme–ligand binding specificity and structural selectivity of CrAT are important for understanding the mechanisms within this enzyme familyCitation2. By catalyzing its reaction in a reversible manner, CrAT regulates the cellular pool of CoA that, in turn, serves as a carrier of activated acetyl groups in the oxidation of energy metabolism substrates and in the synthesis of fatty acids and lipidsCitation3. X-ray crystal structures for both free and L-carnitine bound forms of mouseCitation4 and human peroxisomalCitation5,Citation6 CrAT protein have been solved. Recently, the crystal structure of a ternary complex of CrAT, L-carnitine, and acetyl-CoA has also been publishedCitation7. To date, no biochemical and structural studies have investigated the interactions of CrAT with pharmacological agents that modulate energy metabolism reactions.
Figure 1. Chemical structures of L-carnitine (a), mildronate (3-(2,2,2-trimethylhydrazinium) propionate) (b), and γ-butyrobetaine (GBB) (c).
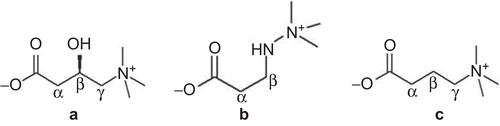
Mildronate (3-(2,2,2-trimethylhydrazinium)propionate) () is a cardioprotective drug that acts as an L-carnitine concentration lowering agent and inhibitor of free fatty acid β-oxidationCitation8,Citation9. Structurally, mildronate is related to L-carnitine and its bioprecursor, γ-butyrobetaine (GBB) (). In mildronate, the γ-carbon of GBB is replaced by a nitrogen atom, which prohibits the enzymatic hydroxylation of the molecule carried out by GBB hydroxylase. Mildronate is known to interact with proteins that are involved in the pathways of the carnitine biosynthesis and transport systems. It is known that mildronate inhibits the biosynthesis of carnitine through GBB hydroxylaseCitation9, carnitine reabsorption in the kidneysCitation10,Citation11, and organic cation/carnitine transporter OCTN2 in the heartCitation12. However, it has also been shown that mildronate inhibits CrAT activity to some extentCitation13. Conversely, it has been reported by several authors that mildronate does not directly inhibit CPT ICitation10,Citation14. Instead, mildronate treatment induces a decrease in carnitine bioavailability for the CPT I catalyzed reaction and, as a result, partially inhibits fatty acid transport into mitochondria and the subsequent production of acetyl-CoA from β-oxidation of fatty acidsCitation9,Citation15. In the presence of low carnitine concentrations and limited availability of acetyl-CoA, the inhibition of CrAT by mildronate might be an important pharmacological mechanism for maintenance of intra-mitochondrial metabolic pathways under ischemic conditions. However, a detailed study of mildronate interactions with the CrAT enzyme has not been performed thus far. Since the regulation of energy metabolism pathways is thought to underlie the cardioprotective mechanism of action of mildronate, this study aimed to characterize the inhibition of the CrAT enzyme. We used biochemical measurements, saturation transfer difference (STD) nuclear magnetic resonance (NMR) spectroscopy, and molecular docking for insights into the binding of mildronate and native ligands to CrAT.
Materials
CrAT from pigeon breast muscle was purchased from Sigma (USA; specific activity 123 U/mgprotein) as a suspension in (NH4)2SO4 solution (3.2 M), potassium phosphate (50 mM), and dithiothreitol (1 mM) at pH 7.0. Prior to NMR studies, the solution was desalted and concentrated into D2O by ultra-filtration through an NMWL (nominal molecular weight limit) 5000 membrane. The enzyme concentration was determined in the desalted solutions by ultraviolet (UV) spectrophotometry, using an extinction coefficient of 70,550 M−1cm−1 based on the number of tryptophans, tyrosines, and disulfide bondsCitation16. L-carnitine and 5,5’-dithiobis-(2-nitrobenzoic acid) (DTNB) were purchased from Acros (Belgium). Acetyl-CoA, 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid sodium salt (HEPES), and ethylenediaminetetraacetate (EDTA) were purchased from Sigma. Mildronate dihydrate was obtained from JSC Grindeks (Latvia).
Methods
CrAT activity
Enzyme activity was determined by following the changes in absorbance at 412 nm using a μQuant™ Microplate Spectrophotometer. The release of CoA-SH from acetyl-CoA was followed using the general thiol reagent DTNB. Assay conditions were DTNB (0.675 mM), HEPES (125 mM), EDTA (2.5 mM), L-carnitine (four concentrations from 0.0625 to 0.5 mM), CrAT (0.3125 U/ml), mildronate (0.3 and 1 mM, water was added for control), and acetyl-CoA (0.1 mM) in a final reaction volume of 200 μL. After addition of mildronate, the reaction mixture was incubated at room temperature for 15 min. The reaction was started by the addition of acetyl-CoA. Immediately after adding acetyl-CoA, the photometric measurements were started, and changes in absorbance were repeatedly measured for 350 s. The enzyme activity unit was defined as nmol CoA-SH released/min. Measurements were repeated three times.
Another set of experiments were carried out to determine IC50 and equilibrium constant for inhibitor binding (Ki) of mildronate under the following assay conditions: DTNB (0.675 mM), HEPES (125 mM), EDTA (2.5 mM), L-carnitine (from 0.0625 to 1 mM), CrAT (0.3125 U/mL), mildronate (seven concentrations from 10 μM to 100 mM), and acetyl-CoA (0.1 mM) in a total reaction volume of 200 μL. The maximum velocity (Vmax), apparent Michaelis constant (Km), IC50, and Ki were calculated using GraphPad Prism 3 software. Double-reciprocal Lineweaver–Burk, Henderson, and Michaelis–Menten kinetic plots were used to display the data.
NMR measurements
All spectra were acquired using a Varian Unity Inova 600 MHz spectrometer equipped with a triple resonance cold probe, and incorporated gradients along the z-axis. Spectra were recorded at 25°C or 1°C (only samples with acetyl-CoA). A one-dimensional (1D) STD pulse sequenceCitation17 with DPFGSE (double-pulsed field-gradient spin-echo) sculpted solvent suppressionCitation18 was used for all STD NMR experiments. STD experiments were recorded with 256–512 scans, an interscan delay of 1.5 s, and 16K data points. Selective saturation of the protein was applied as a train of 40 Gaussian pulses of 50 ms length, separated by a delay of 0.1 ms. The on- and off-resonance saturation was switched between −0.8 and −15 ppm. A spin-lock filter of 10 ms was used to suppress the residual protein signals. All NMR spectra were processed by double-zero filling and multiplication by a squared cosine or Gaussian function prior to Fourier transformation. Spectral processing and analysis was performed using MestRe Nova (Mestrelab Research SL) software.
The samples for standard STD experiments were prepared with CrAT (5 μM) and ligand (mildronate, carnitine, acetyl-CoA (0.25 mM)) in the presence of sodium phosphate (50 mM) buffered 20% D2O/80% H2O (pH 7.0). Samples for competition STD experiments initially contained CrAT (15 μM) and indicator ligand (0.18 mM). For every following experimental step, the concentration of the competing ligand was increased by 0.18 mM.
For determination of the dissociation constant (KD) of the mildronate–CrAT complex (enzyme concentration 20 μM), the 1D solvent-suppressed proton spectra at ligand to protein molar ratios of 1:1; 2:1; 4:1; 6:1; 8:1; 10:1; 15:1, and 20:1 were obtained. The linewidth of the signals of mildronate was estimated by fitting the signals to a Lorentzian lineshape. To compensate for the magnetic field inhomogeneity, the determined linewidths were normalized to the linewidth of 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS) in a particular spectrum. The estimation of KD was accomplished using non-linear least squares fitting in a home written MS Excel workbook kindly provided by Dr. Lee FieldingCitation19.
Molecular docking
Mildronate docking in the active site of CrAT was performed with the Molecular Operating Environment (MOE 2007.09; Chemical Computing Group, Inc., Canada) using built-in methods. The 1.8 å resolution crystal structure of human CrAT complexed with L-carnitine (Protein Data Bank (PDB) identifier 1S5O) was minimized using the CHARMM27 force field, and then used as the model for the macromolecule in docking studiesCitation5. Placement was made using the AlphaPMI algorithm, and placed structures were scored using an Affinity dG scoring functionCitation20. The 100 best-scored structures were finally refined using the CHARMM27 force fieldCitation21,Citation22. From the five lowest-energy structures after refinement, the one that fit best to the STD signal intensities was selected.
After introduction of the M564G mutation, the mutant structure was minimized using the CHARMM27 force field. The docking procedure was as described above.
Results and discussion
Inhibition of CrAT by mildronate
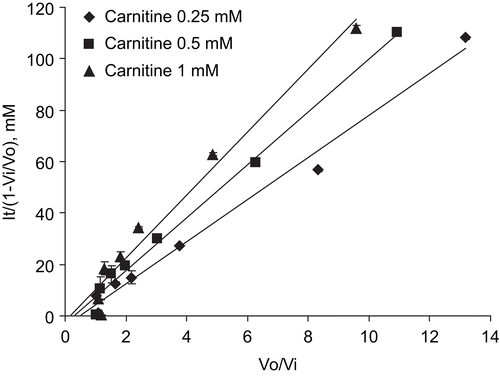
In this study, the CrAT inhibitory potency of the cardioprotective drug mildronate, which is known to inhibit several enzymes and transporters in L-carnitine biosynthesis and transport pathways, was testedCitation23. The effects of mildronate on purified CrAT activity from pigeon breast muscle are presented in , and shows Michaelis–Menten kinetics in the presence of mildronate. First, the kinetic measures of the CrAT reaction were as follows: for carnitine, Km = 0.395 ± 0.107 mM and Vmax = 1.62 ± 0.24 nmol/min. In the presence of 0.3 mM and 1 mM mildronate, the apparent Km values for carnitine were 0.405 ± 0.058 and 0.492 ± 0.116 mM, respectively. The corresponding Vmax value was 1.49 ± 0.03 nmol/min. The double-reciprocal Lineweaver–Burk plots of the obtained data demonstrated the common intersection point close to the vertical axis, thus indicating that mildronate inhibits CrAT in a competitive manner (). The dose–response curves for the inhibition of CrAT by mildronate are shown in . Thus, the calculated Ki and IC50 determined at a fixed carnitine concentration of 0.0625 mM were 1.63 (1.12–1.86) mM and 1.44 (1.26–2.10) mM, respectively; at 0.25 mM were 5.26 (2.99–9.24) mM and 8.59 (4.89–15.10) mM, respectively; at 0.5 mM were 4.65 (3.12–6.93) mM and 10.56 (7.09–15.72) mM, respectively; and at 1 mM were 5.78 (3.83–8.73) mM and 20.44 (13.54–30.87) mM, respectively. These data were analyzed according to HendersonCitation24, and the obtained slopes of Henderson plots () indicated competitive inhibition, which is in agreement with results obtained in the double-reciprocal Lineweaver–Burk plot analysis.
Figure 2. Lineweaver–Burk (A) and Michaelis–Menten (B) kinetic plots for carnitine as the variable substrate at a range of two fixed mildronate concentrations. Points represent mean ± SEM (n = 3).
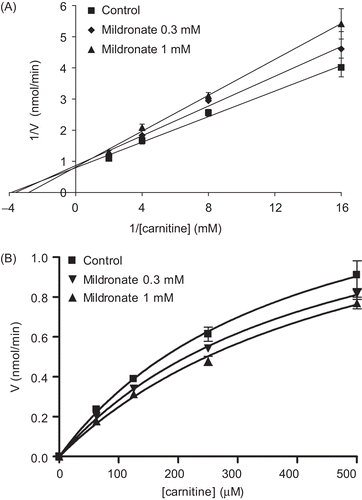
Figure 3. Dose–response curves for the inhibition of carnitine acetyltransferase (CrAT) by mildronate at carnitine concentrations 0.0625, 0.25, 0.5, and 1 mM. Points represent mean ± SEM (n = 3).
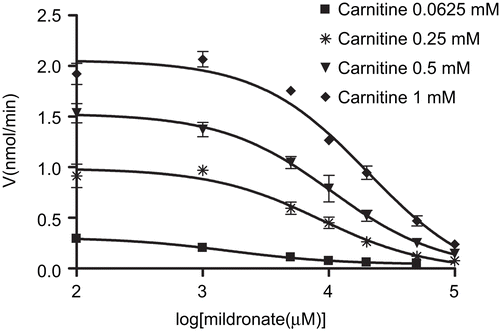
Figure 4. Henderson plot analysis of the mode of interaction of mildronate with CrAT. Points represent mean ± SEM (n = 3). It, total concentration of inhibitor; V0, control velocity; Vi, velocity in presence of inhibitor.
Determination of dissociation constant of the mildronate–CrAT complex
For quantification of the binding affinity of mildronate towards CrAT, an NMR titration was carried out. The NMR methods for the determination of protein–ligand dissociation constants have been recently reviewedCitation25. The NMR linewidths of the signals of mildronate were monitored as a function of the changing solution composition. For fast exchange conditions, the observed NMR linewidth, an approximation to the relaxation rate constant 1/T2, is the mole fraction-weighted average of the free and bound forms. Proton spectra at different CrAT to mildronate ratios were recorded, and the linewidths of the signals were measured. Due to very small linewidth changes during titration (only ~0.5 Hz) for the signals chosen and the inherent sensitivity of the fitting procedure to small errors in the measured linewidth, normalization to an internal reference (DSS) was used to compensate for magnetic field inhomogeneity. The experimental data points were fitted to a simulated binding isotherm by changing the two parameters, KD and νbound. The binding isotherms for the proton signals of mildronate are shown in . The estimated KD was 101 ± 19 μM.
Figure 5. The linewidth of trimethylammonium group (lower), α-methylene group (middle), and β-methylene group (upper) proton signals of mildronate as a function of [mildronate] (mM) at constant [CrAT] = 20 μM. The circles, squares, and triangles are the experimental points, and the curves are the fitted binding isotherms. The estimated KD values are 119 μM (νfree = 1.47 Hz, νbound = 3.85 Hz), 74 μM (νfree = 1.47 Hz, νbound = 4.73 Hz), and 110 μM (νfree = 1.78 Hz, νbound = 5.47 Hz) for trimethylammonium, α-methylene, and β-methylene group signals respectively. The calculated KD for mildronate is 101 ± 19 μM.
![Figure 5. The linewidth of trimethylammonium group (lower), α-methylene group (middle), and β-methylene group (upper) proton signals of mildronate as a function of [mildronate] (mM) at constant [CrAT] = 20 μM. The circles, squares, and triangles are the experimental points, and the curves are the fitted binding isotherms. The estimated KD values are 119 μM (νfree = 1.47 Hz, νbound = 3.85 Hz), 74 μM (νfree = 1.47 Hz, νbound = 4.73 Hz), and 110 μM (νfree = 1.78 Hz, νbound = 5.47 Hz) for trimethylammonium, α-methylene, and β-methylene group signals respectively. The calculated KD for mildronate is 101 ± 19 μM.](/cms/asset/1950b4a2-cb52-4f3c-bb12-d6e45a34860c/ienz_a_383122_f0005_b.gif)
Epitope mapping of mildronate bound to CrAT by STD-NMR
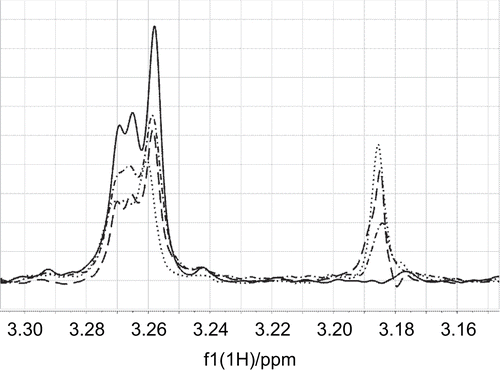
The mildronate binding specificity to the carnitine binding site in CrAT was determined by competition STD-NMR methodCitation26. In the case of two competing ligands, addition of the second ligand to a sample of an indicator ligand and the protein causes disappearance or reduction of the STD signals of the indicator ligand. The competing ligand displaces the indicator ligand in its binding site on the protein; therefore, less magnetization from the bound state of the indicator ligand can be transferred into solution for detection. The extent of signal reduction of the indicator ligand is subject to the binding affinity of the competing ligand. The gradual addition of mildronate to the carnitine–CrAT complex caused a significant and continuous reduction of the STD signals of carnitine and an increase of the mildronate signals (). This showed that mildronate binds to the same binding site as carnitine and suggested a binding constant value in the mM range, similar to that of carnitine.
Figure 6. The γ- methylene proton saturation transfer difference (STD) signal of carnitine at 3.265 ppm was reduced by titration of mildronate to a sample of carnitine (0.18 mM) and CrAT (binding site concentration 15 μM). The STD signal of the trimethylammonium group at 3.185 ppm increased with increasing concentration of mildronate. The lines (——–), (– · – · –), (– – – –), and (· · · · · ·) correspond to carnitine/mildronate ratios of 1:0, 1:1, 1:2, and 1:3.
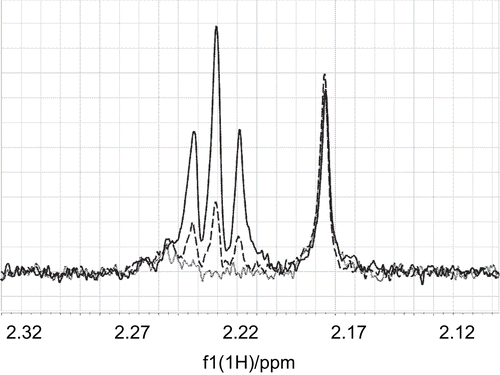
Furthermore, the same principle was used to prove that mildronate does not compete for binding with acetyl-CoA. Experiments with acetyl-CoA were carried out at 25°C and showed its chemical instability at these conditions, indicated by the appearance of two new methyl-resonances over a time of several hours. These changes in the spectrum may be present due to epimerization of one of the chiral carbons. Further experiments with acetyl-CoA were performed at 1°C with a shortened experimental time to reduce the impact of chemical degradation. No reduction of the STD signals of acetyl-CoA upon addition of mildronate to a sample of acetyl-CoA and CrAT was observed, which gives additional evidence that binding of mildronate is specific to the carnitine binding site ().
Figure 7. The intensity of an STD signal of acetyl-CoA at 2.165 ppm did not change upon addition of the non-competing ligand mildronate to a mixture of acetyl-CoA (0.18 mM) and CrAT (15 μM). The STD signal of α-methylene protons at 2.22 ppm increased with increasing concentration of mildronate. The lines (·······), (– – – –), and (——–) correspond to acetyl-CoA/mildronate ratios of 1:0, 1:10, and 1:20.
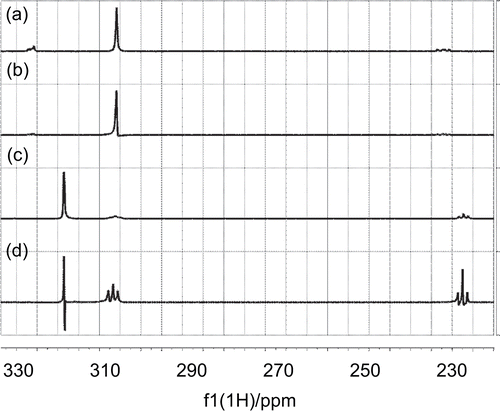
Initial comparison of the STD spectra of mildronate and carnitine () revealed a similar pattern, and suggested the similar conformation for both ligands when bound to CrAT. The STD amplification factors ((I – I0)/I0) were estimated for carnitine γ-CH2 (8.5%), trimethylammonium (37%), and α-CH2 (17%) signals. According to the crystal structure (PDB identifier 1S5O), the γ-CH2 group of carnitine was exposed to the solvent, resulting in the smallest STD amplification factor; meanwhile, the trimethylammonium group was buried in a hydrophobic pocket, giving close contacts to the surface of the protein and resulting in a higher STD amplification factor.
Figure 8. a) 1D 1H NMR spectrum of carnitine; b) 1D STD spectrum of carnitine; c) 1D 1H spectrum of mildronate; d) 1D STD spectrum of mildronate.
Molecular docking
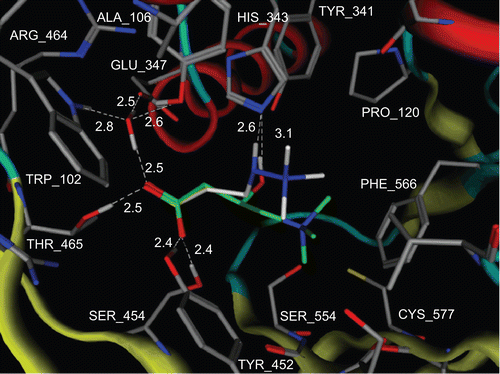
For detailed characterization of the mildronate molecule on CrAT, molecular docking was used, and the results were compared with the STD signal intensities. The STD amplification factors were measured for mildronate trimethylammonium (1%), β-CH2 (17%), and α-CH2 group (11%) signals. The very small STD amplification factor for the trimethylammonium group signal in mildronate as compared to that of carnitine (1% and 37%) suggested conformation with the different position of the trimethylammonium group, possibly exposed to the solvent in the case of mildronate. This conclusion was supported by the docking results showing the conformation, where the trimethylammonium group of the mildronate was exposed to the solvent (). However, the carboxyl and α-CH2 groups of mildronate and carnitine were bound to CrAT very similarly: the carboxylate group has electrostatic interactions with the Arg518 guanidinium group, together with hydrogen-bonding interactions to the side-chain hydrogen donors of Tyr452, Ser454, Thr465, and a water molecule. The α-CH2 groups are in the same position for both ligands, though the STD amplification factor for the carnitine one was larger due to better binding.
The catalytic His343 is hydrogen-bonded to the NH group of mildronate, instead of the OH moiety in carnitine. This caused differences in binding of both ligands. The trimethylammonium group of mildronate was situated in a channel approximately 4 å away from the protein surface and was exposed to the solvent, whereas the trimethylammonium group of carnitine was buried in a hydrophobic pocket. The lack of hydrophobic interactions between the trimethylammonium group and the protein likely accounted for the weaker binding of mildronate to CrAT as compared to carnitine. This observation is in agreement with results obtained in biochemical measurements of CrAT activity, where Km for carnitine was estimated to be three times lower than Ki for mildronate ( and ).
In earlier studies it was demonstrated that mildronate does not inhibit CPT ICitation10,Citation14. However, in the case of CrAT, our results clearly show that mildronate exhibits a binding mode similar to carnitine and therefore could also bind to other carnitine acyltransferases. Since the M564G mutant of CrAT was characterized recently and has been shown to exhibit substrate specificity close to carnitine octanoyltransferase (CrOT; EC 2.3.1.137) and CPT ICitation27, we performed molecular docking studies on this mutant model (data not shown). The obtained results for carnitine and mildronate were identical to those obtained on native CrAT. This can be explained by the fact that in the M564G mutant the binding site of carnitine and mildronate remains unchanged due to the large distance between the active site and the mutated amino acid (the closest distance between any two atoms of carnitine and mutated G564 is 9.9 å). Our results show that the specificity of mildronate binding does not depend on differences in size of the acyl-group binding pocket in acyltransferases. Instead, the mildronate molecule is able to discriminate between the carnitine binding sites of CrAT (present study) and CPT ICitation10,Citation14, which might bring about pharmacological consequences.
The obtained results give evidence that the CrAT enzyme might be considered as a molecular target for the cardioprotective activity of mildronate. The obtained values of the half-saturation constant and IC50 of mildronate for CrAT inhibition were close to the concentrations that can be achieved in rat heart and liver after long-term administration of a pharmacologically relevant dose of the drug. Thus, the concentration of mildronate after 1 month of treatment (100 mg/kg daily) in rat heart and liver reached 640 ± 48 nmol/g and 1480 ± 223 nmol/g, respectively (Dr. O. Pugovich, unpublished observation). Through a mild inhibitory effect on CrAT, mildronate might regulate the cellular pool of coenzyme A and availability of activated acetyl groups in a way that is favorable under ischemic conditions.
Figure 9. Proposed model for the positioning of carnitine and mildronate in CrAT enzyme. Carnitine carbons are shown in green and mildronate carbons are shown in white. All hydrogens, except those involved in hydrogen bonds, are omitted. Protein backbone is shown as tubes. Hydrogen bonds are shown as white dashed lines. White labels correspond to amino acid residues and distances between heavy atoms, which are involved in hydrogen bonding. Produced with MOE 2007.09.
Conclusions
In conclusion, our results show that mildronate inhibits CrAT in a competitive manner through binding to CrAT, which occurs at the carnitine binding site. The bound conformation of mildronate closely resembles that of carnitine except for the orientation of the trimethylammonium group, which in the mildronate molecule is exposed to the solvent. The dissociation constant of mildronate and CrAT complex is approximately 0.1 mM, and the Ki is 1.63 mM. The obtained results suggest that the inhibition of CrAT occurs at pharmacologically relevant concentrations and the cardioprotective effect of mildronate might be partially mediated by CrAT inhibition and following regulation of cellular energy metabolism pathways.
Acknowledgements
We would like to thank Dr. L. Fielding (Organon Laboratories Ltd, UK) for assistance with binding curve fitting procedures.
Declaration of interest: This study was supported by a grant from the Latvian Science Council (05.1461) and Latvian State Research Program “New medicines and biocorrection tools: design, transport forms and mechanisms of action.”
References
- Ramsay RR, Zammit VA. Mol Aspects Med 2004;25:475–93.
- Ramsay RR, Naismith JH. Trends Biochem Sci 2003;28:343–6.
- Ramsay RR, Arduini A. Arch Biochem Biophys 1993;302:307–14.
- Jogl G, Tong L. Cell 2003;112:113–22.
- Govindasamy L, Kukar T, Lian W, Pedersen B, Gu Y, Agbandje-McKenna M, et al. J Struct Biol 2004;146:416–24.
- Wu D, Govindasamy L, Lian W, Gu Y, Kukar T, Agbandje-McKenna M, et al. J Biol Chem 2003;278:13159–65.
- Hsiao YS, Jogl G, Tong L. J Biol Chem 2006;281:28480–7.
- Liepinsh E, Vilskersts R, Loca D, Kirjanova O, Pugovichs O, Kalvinsh I, et al. J Cardiovasc Pharmacol 2006;48:314–19.
- Simkhovich BZ, Shutenko ZV, Meirena DV, Khagi KB, Mezapuke RJ, Molodchina TN, Kalvins IJ, Lukevics E. Biochem Pharmacol 1988;37:195–202.
- Kuwajima M, Harashima H, Hayashi M, Ise S, Sei M, Lu K, Kiwada H, Sugiyama Y, Shima K. J Pharmacol Exp Ther 1999;289:93–102.
- Spaniol M, Brooks H, Auer L, Zimmermann A, Solioz M, Stieger B, et al. Eur J Biochem 2001;268:1876–87.
- Grube M, Meyer zu Schwabedissen HE, Prager D, Haney J, Moritz KU, Meissner K, et al. Circulation 2006;113:1114–22.
- Shutenko Z, Simkhovich BZ, Meirena DV, Kalvin’sh II, Lukevits EI. Vopr Med Khim 1989;35:59–64.
- Tsoko M, Beauseigneur F, Gresti J, Niot I, Demarquoy J, Boichot J, et al. Biochem Pharmacol 1995;49:1403–10.
- Spaniol M, Kaufmann P, Beier K, Wuthrich J, Torok M, Scharnagl H, et al. J Lipid Res 2003;44:144–53.
- Gill SC, von Hippel PH. Anal Biochem 1989;182:319–26.
- Mayer M, Meyer B. J Am Chem Soc 2001;123:6108–17.
- Hwang TL, Shaka AJ. J Magn Reson 1995;112:275–279.
- Fielding L, Rutherford S, Fletcher D. Magn Reson Chem 2005;43: 463–70.
- Bruno IJ, Cole JC, Lommerse JP, Rowland RS, Taylor R, Verdonk ML. J Comput Aided Mol Des 1997;11:525–37.
- Goodford PJ. J Med Chem 1985;28:849–57.
- Muegge I, Martin YC. J Med Chem 1999;42:791–804.
- Dambrova M, Liepinsh E, Kalvinsh I. Trends Cardiovasc Med 2002;12:275–9.
- Henderson PJ. Biochem J 1972;127:321–33.
- Fielding L. Progr Nucl Magn Reson Spectrosc 2007;51:219–242.
- Wang YS, Liu D, Wyss DF. Magn Reson Chem 2004;42:485–9.
- Cordente AG, Lopez-Vinas E, Vazquez MI, Swiegers JH, Pretorius IS, Gomez-Puertas P, et al. J Biol Chem 2004;279:33899–908.