Abstract
In drug discovery, different methods exist to create new inhibitors possessing satisfactory biological activity. The multisubstrate adduct inhibitor (MAI) approach is one of these methods, which consists of a covalent combination between analogs of the substrate and the cofactor or of the multiple substrates used by the target enzyme. Adopted as the first line of investigation for many enzymes, this method has brought insights into the enzymatic mechanism, structure, and inhibitory requirements. In this review, the MAI approach, applied to different classes of enzyme, is reported from the point of view of biological activity.
Table of contents
Introduction 1292
1. Lyases 1292
1.1. Ornithine decarboxylase 1292
1.2. Porphobilinogen synthase 1292
2. Ligase 1293
2.1. Adenylosuccinate synthase 1293
2.2. Acetyl-CoA carboxylase 1293
2.3. Salicyl-AMP ligase 1293
3. Oxidoreductase 1293
3.1. Dopamine β-hydroxylase 1293
3.2. 4-Hydroxybenzoate 3-monooxygenase 1295
4. Transferases 1296
4.1. 3-Deoxy-d-manno-2-octulosonate-8-phosphate (KDO8P) synthase 1296
4.2. Spermidine synthase and spermine transferase 1296
4.3. Glycinamide ribotide transformylase, 5-amino-4-imidazol carboxyamide ribotide transformylase, and thymidylate synthase 1296
4.4. Protein kinases 1297
4.5. Purine nucleoside phosphorylase 1302
4.6. Methyl transferases 1302
4.7. Thymidine phosphorylase 1305
4.8. Glycosyltransferases 1306
4.9. Acetyltransferases 1308
4.10. Farnesyl/geranylgeranyl transferases 1312
4.11. Riboflavin synthase 1314
4.12. Nicotinamide mononucleotide adenyltransferase 1315
Conclusion 1315
Acknowledgements 1315
References 1315
Introduction
One of the most important aspects of drug design is for small molecules to achieve high specificity and efficacy toward their given biological target, whether it is a membrane-bound receptor or an enzyme. For lead compound identification based on enzyme inhibition, several methods have been developed to enhance specificity and potency. Such compounds are transition-state analogs, suicide or mechanism-based inhibitors, and multisubstrate adduct inhibitors (MAIs)Citation1. The design of transition-state analogs requires a precise understanding of the enzyme mechanism and of its transition-state/enzyme complex (E–S). Alternatively, suicide or mechanism-based inhibitors require, for inhibition, the molecule to interact with the target enzyme in such a way as to initiate a catalytic process, thus resulting in the formation of a stable inhibitor–enzyme complex. The MAI approach can potentially be applied to all enzymatic reactions in which at least two molecules (cofactor included) are simultaneously present and reacting in the enzyme active site. As such, the combination of structural features taken from each reagent into a single molecule potentially increases the binding efficacy as order is introduced, and the binding specificity due to the substrate/cofactor synergistic effect on recognition patterns. This combination of several substrates involved in an enzymatic reaction was termed by Wolfenden as multisubstrateCitation2. Currently, the design of MAIs is one of the best means to obtain mechanistic and structural information on an enzyme and to create new inhibitors with potentially high potency.
Often the use of a cofactor mimic moiety allows for an increase in affinity toward the enzyme, while analogs of the substrate that compete with the natural substrate in the active site enhance the specificity. However, two important aspects in the MAI approach have to be considered: the points of attachment of the linker between the cofactor and the substrate and the length of this linker, which must allow for binding to both substrate and cofactor pockets. We will now present developments that have been made in drug design through the MAI approach by examining the different classes of enzyme and the approach to their inhibition.
1. Lyases
1.1. Ornithine decarboxylase
One of the first studies on MAIs described specific inhibitors for prokaryotic and eukaryotic ornithine decarboxylase (EC 4.1.1.17)Citation3. Ornithine decarboxylase (ODC) catalyzes the decarboxylation of ornithine, producing the diamine putrescine. ODC utilizes pyridoxal-5′-phosphate (PLP) as the cofactor and ornithine or lysine as the substrate. Coward et al. synthesized the reduced Schiff base adduct analogous to the Schiff base involved in enzyme-catalyzed decarboxylation ().
Table 1. Effect of adduct 1 at various concentrations of pyridoxal-5′-phosphate (PLP).
The reduced Schiff base adducts 1a–c were tested as inhibitors of several decarboxylases and aminotransferases. It was found that adducts 1a and 1c were both efficient against ornithine and lysine decarboxylase. In the same manner, it was shown that apo-dopa decarboxylase (EC 4.1.1.28) was inhibited by both N-(5′-phosphopyridoxyl)tyrosine and N-(5′-phosphopyridoxyl)phenylalanine, while other PLP–amine adducts were much less effective.
1.2. Porphobilinogen synthase
Porphobilinogen synthase (PBGS) (5-aminolevulinate dehydratase (ALAD); EC 4.2.1.24) catalyzes the condensation of two molecules of 5-aminolevulinic acid (ALA; ) to produce porphobilinogen (PBG), which is an intermediate in the biosynthesis of tetrapyrrolic natural products such as porphyrins and chlorophylls Citation4. In an aim to increase the knowledge of the active site of the enzyme and to elucidate its mechanism, Neier et al. developed bisubstrate analogs () which incorporated two γ-keto carboxylic groups for recognition with the enzyme’s active site, and were attached together by several linkersCitation5.
Scheme 1. Bisubstrate analogs for porphobilinogen synthase.
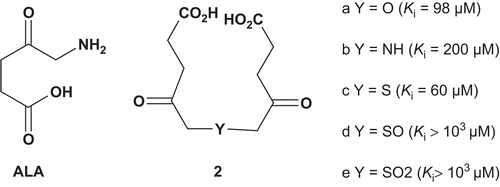
These bisubstrates were evaluated as potential inhibitors of Mg2+-dependent PBGS from Pseudomonas aeruginosa (ALA Km = 0.33 mM). It appeared that bisubstrates with a small linker (a–c) were about 500-fold more active than were the two others. The poor affinity of the compounds d and e toward the enzyme could be explained by the size of the linkers sulfoxide and sulfone, which are bigger than amine or thio linkersCitation5.
2. Ligase
2.1. Adenylosuccinate synthase
Adenylosuccinate synthase (AdSS; EC 6.3.4.4) catalyzes the first step in the transformation of inosine monophosphate (IMP) into adenosine monophosphate (AMP)Citation6. Hydantocidin, known as pro-herbicide, is first converted into hydantocidin monophosphate (HMP), and this molecule mimics either IMP or AMP. Evidence from crystallographic studies of the enzyme showed that hydantocidin replaced IMP in the active siteCitation7–10. Hadacidin is also a natural substrate of AdSS, which was reported as a competitive inhibitor of aspartic acid but affecting the enzyme at a different siteCitation11. Based on the crystal structure of the complex AdSS–hadacidin, Hanessian et al. synthesized two bisubstrate hybrids composed of a covalent attachment between HMP and hadacidin ()Citation12.
Scheme 2. Bisubstrate hybrids hydantocidin monophosphate (HMP)/hadacidin for adenylosuccinate synthase.
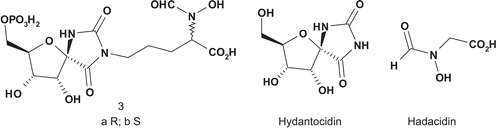
These two adducts were evaluated as inhibitors of AdSS from E. coli and wheat (IMP Km = 0.041 mM), with 3b displaying an IC50 of 0.043 μM for AdSS from E. coli and 0.200 μM for AdSS from wheat.
The inhibitor 3b was 10- and 100-fold more potent than HMP and hadacidin respectively. The linking of two relatively weak active inhibitors into a single molecule such as 3b enhanced significantly the enzymatic inhibitory activity as well as specificity when compared to the individual natural substratesCitation12.
2.2. Acetyl-CoA carboxylase
Acetyl-CoA carboxylase (ACC; EC 6.3.4.9) catalyzes the biotin-dependent carboxylation of acetyl-CoA to produce malonyl-CoA. ACC has been connected to obesity, type 2 diabetes, and microbial infectionCitation13–16. This makes ACC a target of choice for the development of new drugs. Based on a steady-state kinetic study, Waldrop synthesized and evaluated a unique bisubstrate analog as inhibitor of ACC () featured by the coenzyme-A linked to the chloroacylated biotin analog via an acyl bridgeCitation17,Citation18.
Scheme 3. Bisubstrate analogs for acetyl-CoA carboxylase.
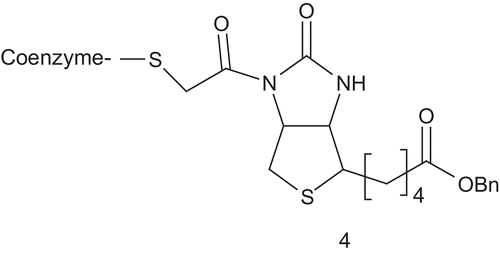
Using malonyl-CoA (Km = 0.1 mM) as a variable substrate, 4 showed competitive inhibition, with a Ki value of 23 ± 2 μM. In contrast, when biotycin (Km = 8.25 mM) was applied as the variable substrate, non-competitive inhibition was observedCitation18.
2.3. Salicyl-AMP ligase
The elucidation of the mycobactrin biosynthetic pathway showed that salicyl-AMP (MbtA) is involved in the initiation of mycobactrin chain growth. Using this finding, Aldrich et al. developed new bisubstrate analog inhibitors of salicyl-AMP, with an acylsulfamate linkage mimicking the acyl-adenylate intermediate ()Citation19–25.
Scheme 4. Bisubstrate analogs for salicyl-AMP.
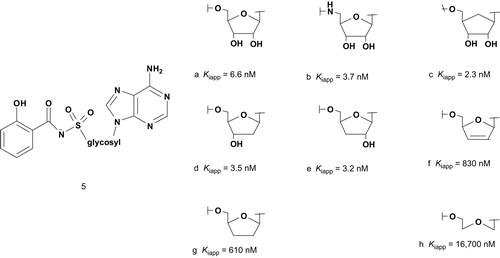
Biochemical analysis of MbtA using bisubstrate inhibitor has provided details of the reaction mechanism. With these results, Aldrich et al. provided information for future structure–activity relationship (SAR) studies and moreover were able to identify several modifications to the glycosyl inhibitors template, in particular on the nucleobase part, which improved the inhibitory activityCitation26. Species specificity was also evidenced; quite noticeable is the identification of novel compounds as potent inhibitor (5b) of M. tuberculosis growth such as isoniazid, the most commonly prescribed anti-tuberculosis drug.
3. Oxidoreductase
3.1. Dopamine β-hydroxylase
Dopamine β-hydroxylase (DβH; EC 1.14.17.1) is a tetrameric, copper containing, mixed-function oxidase that catalyzes benzylic hydroxylation of dopamine to (R)-norepinephrine in the sympathetic nervous systemCitation27. Kruse et al. hypothesized that the DβH-catalyzed oxidation involved simultaneous binding of oxygen to a copper(I) site and to the phenylethylamine near the aromatic binding site. Thus, they developed new multisubstrate inhibitors ()Citation28. In this first study, a phenyl or 4-oxygenated phenyl group was used as a mimic of the dopamine catecholic nucleus for recognition by the aromatic binding site of the enzyme. Synthesis and testing of a first panel of inhibitors was undertaken in order to optimize the space between the aryl system and the dopamine catechol mimicCitation28. Compound 6 (X = OH, Y = CH2, IC50 = 2.6 μM, Ki = 0.00549 μM at pH 4.5, 0.344 μM at pH 6.6) binds DβH approximately 105-fold more tightly than the tyramine (Km = 5.65 mM) and appears more potent than furasic acid, the inhibitor which underwent clinical trials.
Scheme 5. Multisubstrate adduct for dopamine β-hydroxylase.
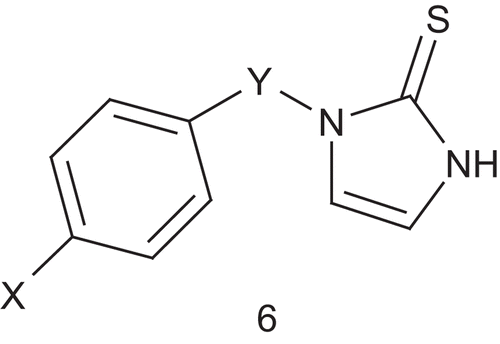
The same group reported a SAR study on 1-benzylimidazole-2-thione and its derivatives ()Citation29. This allowed identification of the inhibitor’s metabolic liability and determination of whether the affinity of multisubstrate inhibitors to DβH occurs by direct binding to the phenethylamine substrate site. Another series of derivatives were then prepared and some more potent inhibitors were revealedCitation30. The most potent inhibitor (6: X = 3,5-F2, 4-OH, Y = CH2, IC50 = 0.074 μM) was binding the enzyme approximately 106-fold more tightly than tyramine. However, a lack of oral bioavailability was also identified, and this was associated with the phenolic group, which is a good site for metabolic conjugation in vivo. Fortunately, the 3,5-dihalo-substitution pattern was a viable replacement for the metabolically liable 4-hydroxyl groupCitation29. In order to improve the potency of the previous inhibitor, Berkowitz et al. reported a new approach with substituted pyridyl moieties as isostere of the aryl groups present in substrates such as phenethylamine and p-tyramine and in the 1-(arylmethyl)imidazole-2-thiones ()Citation31.
Scheme 6. Modified multisubstrate adducts for dopamine β-hydroxylase.
The compounds 7a–c and 8, 9, 10 were tested for inhibition of DβH in vitro and in vivo (), with reference data for three 1-(arylmethyl)-imidazole-2-thionesCitation28–30 and a standard DβH inhibitor (furasic acid)Citation32. Compounds 7a–c were potent as DβH inhibitors in vitro (IC50 ≈ 10−4 M) and even more potent in vivo. Particularly for compound 7a (2-pyridyl), the in vivo effects were comparable to those of furasic acid. Of the oxy-substituted compounds 8–10, only the 5-hydroxy-2-pyridylmethyl compound (8) was efficient against DβH (IC50 ≈ 10−5 M) in vitro, but unfortunately it had a weak antihypertensive effect in vivo.
Table 2. DβH inhibition DA/NE ratioa and antihypertensive activities of 1-(pyridylmethyl)imidazole-2-thione.
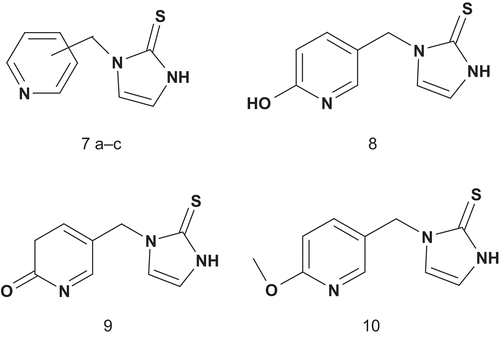
The same group then investigated the possibility of enhancing inhibitor potency by modifying the copper-binding portion of the inhibitors in order to obtain further information about the enzyme copper binding siteCitation33. These results showed that the soft sulfur was clearly required for optimal activity. After having established the sulfur requirement, the investigation moved toward variation of the ligand ring. A series of DβH alkylaryl-substituted heterocycles were synthesized and tested for inhibition of DβH. The data showed weak affinitiesCitation34. A chiral approach was reported for inhibition of DβH. The series of novel potent, rigid, and chiral bisubstrate inhibitors was designed to mimic phenylethylamine and dioxygen substrates in order to study the electronic, steric, and proximity constraints of the DβH active siteCitation27. These compounds were tested, and the results indicated that the DβH active site possessed a steric and electronic tolerance toward these inhibitors, allowing for the interaction of the enantiomeric pairs of inhibitors with the enzyme especially in the region of the two copper centers. It should be emphasized that 11a and 11b () were found to be potent competitive inhibitors of DβH.
Scheme 7. Rigid and chiral bisubstrates of dopamine β-hydroxylase.
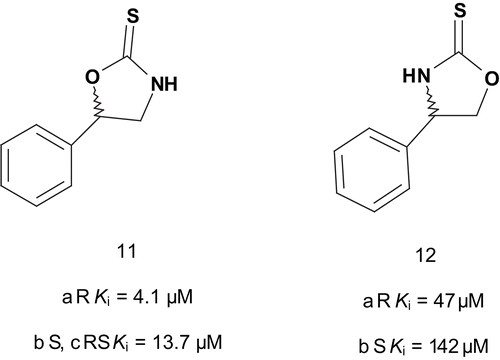
In addition, the transposition of the oxygen and nitrogen in the thiooxazolidone ring of 11a and 11b by displacement of 5-phenyl to 4-phenyl (compounds 12a and 12b) decreased strongly the inhibition potency.
3.2. 4-Hydroxybenzoate 3-monooxygenase
4-Hydroxybenzoate 3-monooxygenase (PHBH; EC 1.14.13.2) is well known as the NADPH-dependent enzyme flavin-monooxygenase. The enzyme mechanism has been characterized as a random sequential addition of the two substrates p-hydroxybenzoic acid (Km = 21 μM) and NADPH (Km = 57 μM) to the enzyme and oxidized flavin cofactorCitation34.
Salituro et al. reported the design of new multisubstrate inhibitors in which the NADPH mimic was simplified by an unsubstituted phenyl ring, to stabilize the π–π interaction between the aromatic ring of this group and the flavin nucleusCitation34. In addition, a methyleneoxy moiety was used as a linker between the p-hydrobenzoic acid and the NADPH mimic ().
Scheme 8. Multisubstrates for 4-hydroxybenzoate 3-monooxygenase.
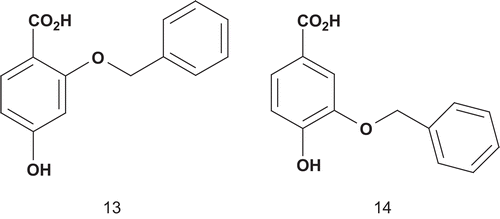
Both potential inhibitors were initially evaluated by modeling experiments, which showed that 13 was stacked under the flavin’s B ring with a similar position to the p-hydrobenzoate substrate, whereas 14 moved out of the plane. This suggested that 13 would be the best potential inhibitor due to the favorable π–π interactionsCitation34. Then, both compounds were tested as inhibitors of PHBH and, in agreement with the modeling predictions, 13 had the highest affinity (Ki = 59 nM) for the enzyme.
4. Transferases
4.1. 3-Deoxy-d-manno-2-octulosonate-8-phosphate (KDO8P) synthase
KDO8P synthase (EC 2.5.1.55) catalyzes the condensation reaction between d-arabinose-5-phosphate (A5P) and phosphoenolpyruvate (PEP), forming KDO8P and releasing an inorganic phosphateCitation35.
Du et al. reported the synthesis of the first bisubstrate inhibitor of KDO8P synthase, which is crucial in the assembly process of the lipopolysaccharides of most gram- negative bacteriaCitation36–38. According to studies carried out on the enzyme’s mechanism, A5P and an amino PEP analog could be combined () to give access to 15Citation39–43.
Scheme 9. Bisubstrate for 3-deoxy-d-manno-2-octulosonate-8- phosphate (KDO8P) synthase.
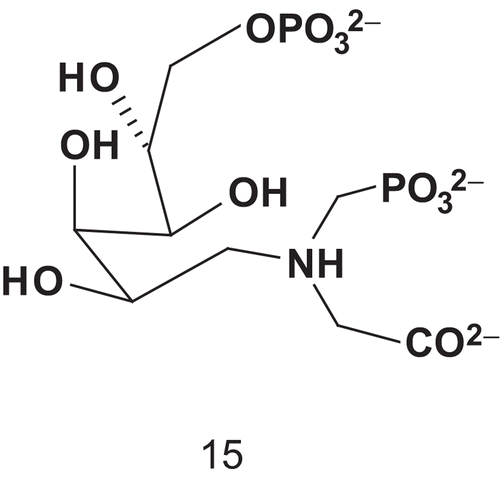
Compound 15 was evaluated as an inhibitor of KDO8P synthase to obtain insights into the enzyme mechanism. Bisubstrate 15 appeared to bind the enzyme 1500-fold more tightly than KDO8P and 20-fold more tightly than PEPCitation36. The study showed that 15 functioned as a slow-binding inhibitor with a dissociation constant of 0.42 μM (Km (PEP) = 8 μM).
4.2. Spermidine synthase and spermine transferase
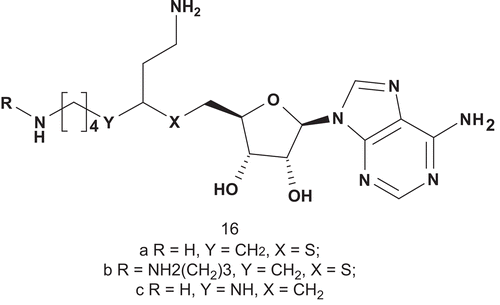
The biosynthesis of polyamines is carried out by three highly conserved polyamine biosynthetic enzymes: ornithine decarboxylase, putrescine aminopropyltransferase (PAPT), and spermidine aminopropyltransferase (SAPT). Inhibition of polyamine biosynthesis has potential as chemotherapy and as antiproliferative therapyCitation44,Citation45. A series of aminopropyltransferase inhibitors have been developed for spermidine synthase (PAPT; EC 2.5.1.16) and spermine transferase (SAPT; EC 2.5.1.22)Citation46. It has been demonstrated, in the case of spermidine synthase from E. coli, that transfer of the aminopropyl group occurs via a ternary complex by direct nucleophilic attackCitation47. Synthesis and biological evaluation of S-adenosyl-1,8-diamino-3-thiooctane (AdoDATO; 16a) showed that this compound was the most potent inhibitor known for spermidine synthaseCitation46,Citation47. This high potency of inhibition was due to the fact that AdoDATO was a multisubstrate analog of the transition stateCitation48. From this preliminary study, S-adenosyl-1,12-diamino-3-thio-9-azadodecane (AdoDATAD; 16b) was synthesized and biologically evaluated as the corresponding multisubstrate adduct inhibitor for the spermine synthase reaction. AdoDATO provided the structural basis for the design of 3-(R,S)-(5′-deoxy-5′-carbadenos-6′-yl) spermidine (AdoSpd; 16c). This inhibitor displayed good inhibition of putrescine aminopropyltransferase (PAPT)Citation49. Nevertheless, AdoSpd was less selective than AdoDATO, and inhibited spermidine aminopropyltransferase (SAPT) to a significant extent ().
Scheme 10. Multisubstrate adducts for spermine synthase reaction.
4.3. Glycinamide ribotide transformylase, 5-amino-4-imidazol carboxyamide ribotide transformylase, and thymidylate synthase
4.3.1. Glycinamide ribotide transformylase
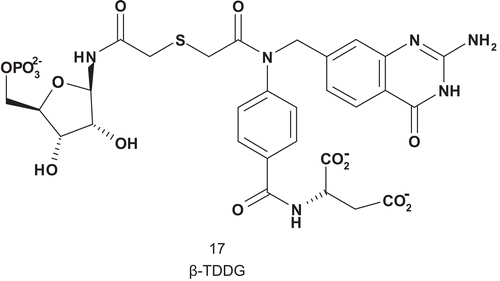
Glycinamide ribotide transformylase (GAR-Tase; EC 2.1.2.2) catalyzes the first step in the de novo purine biosynthesis pathway requiring folate as a cofactorCitation50. A physiological effect was obtained with 5,10-dideaza-5,6,7,8-tetrahydrofolic acid against solid tumor and this was attributed to the inhibition of GAR-TaseCitation51. Benkovic et al. described the first successful MAI (β-TDDG) for GAR-Tase with a very good affinity for the enzyme, with a Ki value three-fold higher than that of the substrate (Km (GAR) = 23 μM) ()Citation52. β-TDDG acted as a slow, tight-binding inhibitor against four species of GAR-Tase (E. coli, Avian, Hela O, and L1210). In addition, β-TDDG was the most potent inhibitor reported for this enzyme: Kd = 250 ± 50 nM. Using this compound, Wilson solved the crystal structure of the GAR-Tase active siteCitation53–55.
Scheme 11. Multisubstrate for glycinamide ribotide transformylase (GAR-Tase).
4.3.2. 5-Amino-4-imidazol carboxyamide ribotide transformylase (AICAR-Tase)
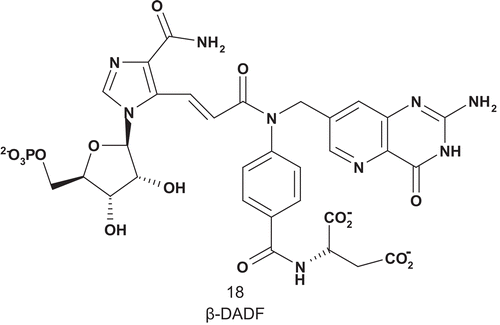
Benkovic also developed the first MAI for AICAR-Tase (EC 2.1.2.3), β-DADF (18) (), including both substrates used by the enzymeCitation56. β-DADF was evaluated as an inhibitor of AICAR-Tase, with a Kd of 20 nM. β-DADF was bound 10Citation3-fold more tightly than each of the other substrates by the transferase, with complete selectivity for the AICAR enzyme over GAR-TaseCitation57.
Scheme 12. Multisubstrate for amino-4-imidazol carboxyamide ribotide transformylase (AICAR-Tase).
4.3.3. Thymidylate synthase
The concept of “thymineless death” has for many years been used to justify thymidylate synthase inhibition (TS; EC 2.1.1.45) as a target for cancer chemotherapy, as demonstrated by Koyama et al.Citation58–60.
Mertes et al. synthesized the first thymidylate synthase inhibitor with a Ki of 0.75 μM, which was a thymidylate substituted on the 5-methyl with a simple tetrahydroquinoxalineCitation61. Subsequent to this, Broom et al. described the synthesis and biological evaluation of other multisubstrate analog inhibitors ()Citation59.
Table 3. Inhibition of human thymidylate synthase.
Compound 19b proved to be a potent competitive inhibitor of TS. Broom et al. then focused on the synthesis of bisubstrate analogs that would retain the binding abilities of 19b and have greater flexibility, but without the chiral carbonCitation62. Unfortunately, these new bisubstrates were less potent inhibitors than 19b. The hypothesis was that the 6-position of the pyrimidine might undergo attack by a sulfhydryl group present in the active site in order to create a ternary complex resembling 19b. This last study suggested that these inhibitors fitted the active site, but might act more as product-substrate analogs than as -bisubstrate analogsCitation62.
Recently, Lebioda et al. reported cooperative inhibition of hTS exploiting both active site and allosteric inhibitions, creating synergy by using two inhibitors binding two different subunits of TSCitation62. The concept developed here to target two non-equivalent sites may alleviate the development of drug resistance in patients, even though the resistance through increased expression cannot yet be addressed by this approach.
4.4. Protein kinases
Kinases catalyze the transfer of the γ-phosphate of nucleoside triphosphates (usually adenosine triphosphate; ATP) to a functional group on an acceptor molecule. X-ray crystallography and nuclear magnetic resonance (NMR) analysis have been particularly useful in helping to define the nature of the binding sitesCitation64. The progress in this field has shown that such inhibitors could be invaluable as biological reagents and serve as therapeutically useful compounds for the treatment of a wide variety of diseasesCitation64.
4.4.1. Tyrosine kinase
According to Holden, the MAI approach for protein kinases (PKs) is only useful with enzymes that directly phosphorylate their substrate without formation of a phosphorylated enzyme intermediate. As a result of studies into other classes of kinases and Goldberg’s and Wong’s investigations, it is now possible to assume that tyrosine phosphorylation of angiotensin II proceeds via a ternary complex, thus amenable to inhibition by multisubstrate analogsCitation65.
Based on this, Holden et al. adopted the MAI approach to probe the distance between the ATP and tyrosine binding sites in a tyrosine-specific protein kinase ()Citation66.
Scheme 13. Multisubstrate for tyrosine kinase.
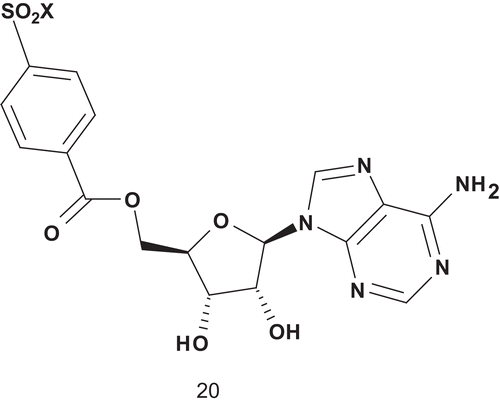
Poor inhibition was achieved with compound 20, and as such they subsequently reported the synthesis and evaluation of multisubstrate inhibitors containing a polyphosphate linkage between the tyrosine mimic block and the adenosine ()Citation66. However, the improvement of inhibitory activity was not really significant.
Table 4. Inhibition of tyrosine kinase.
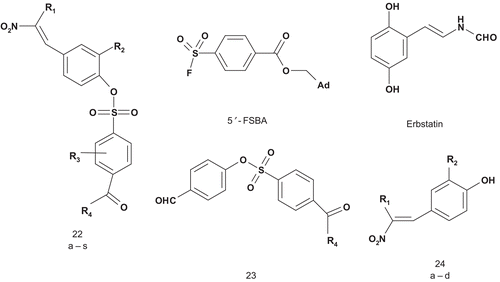
Traxler et al. considered the multisubstrate inhibitor approach to develop potential bisubstrate inhibitors of the epidermal growth factor (EGF)-receptor tyrosine kinaseCitation67. Their strategy was to combine both elements of ATP and tyrosine. Therefore, 5′-[4-(fluorosulfonyl)benzoyl]adenosine (5′-FSBA) was covalently attached to tyrosine mimics via a sulfonyl moiety ()Citation68–71. Compounds were tested as selective inhibitors of EGF-receptor (EGF-R) tyrosine. Compounds 23–24 showed a moderate inhibitory activity for EGF-R tyrosine kinase. Their IC50 values were similar to that of erbstatinCitation72. However, these compounds were selective for EGF-R tyrosine kinase. A stronger inhibition was observed for compounds 22a–s, with Ki values of about 1 μM. The most potent inhibitor (R1 = H; R2 = H; R3 = 2-OH; R4 = OH) showed a Ki value of 0.054 μM. Additionally, it offered high selectivity for EGF-R tyrosine kinase with respect to v-abl tyrosine kinase and protein kinase C (PKC)Citation67.
Scheme 14. Bisubstrates for epidermal growth factor (EGF)-receptor tyrosine kinase.
Recent investigations into the design of inhibitors for PKs led Uri’s group to develop nanomolar bisubstrate analog inhibitors of basophilic PKsCitation64. Their strategy aimed to combine an oligoarginine peptide with adenosine, adenosine-5′-carboxylic acid, and 5-isoquinolinesulfonic acid ().
Scheme 15. Bisubstrate analogs for basophilic protein kinases (PKs).
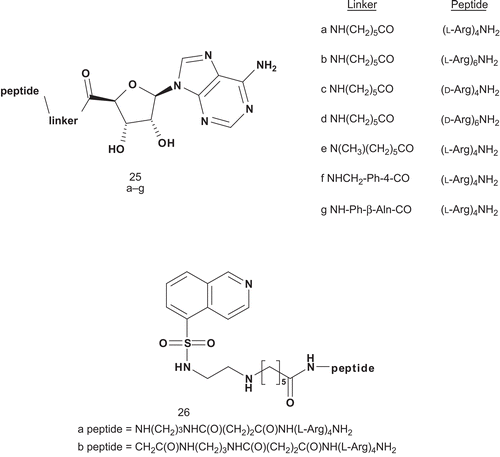
All these compounds have been evaluated as inhibitors of cAPK (calmodulin-dependent protein kinase), and 25d and 25b were found to be the most potent inhibitors with an IC50 of 8.3 nM and 5.3 nM, respectively.
4.4.2. Adenylate kinase
Adenylate kinase (AK; EC 2.7.4.3) was first investigated with the MAI approach by Wolfenden and Lienhard. The latter used this strategy to develop a potent inhibitor () of AKCitation73. This enzyme is a phosphotransferase enzyme that catalyzes the reversible transfer of the terminal phosphate group between ATP-Mg2+ and adenosine monophosphate (AMP) to provide two adenosine diphosphates (ADPs)Citation74.
Scheme 16. Multisubstrate for adenylate kinase.
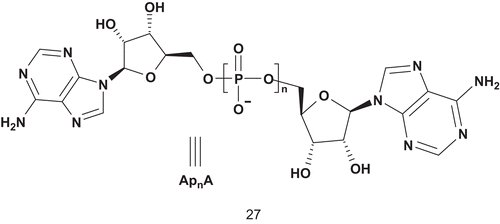
Thus, Lienhard synthesized the first series of MAIs (n = 2, 3, 4, 5), and their biological evaluation led to the identification of a potent inhibitor of adenylate kinase, Ap5A, which was able to inhibit the reaction of adenylate kinase by 55% at 3 × 10−8 M. At the same time, Lienhard demonstrated that requirements for potent inhibition were that two adenosine groups needed to be linked by a polyphosphate bridge containing at least five phosphoryl groupsCitation73.
4.4.3. Thymidine kinase and thymidylate kinase
Thymidine kinase (TK; EC 2.7.1.21) plays a central role in the nucleotide salvage pathway. It catalyzes the transfer of the γ-phosphoryl group of ATP to thymidine to produce thymidine monophosphate (TMP). It is an essential enzyme for cell proliferation, and thus an attractive target for the development of drugs against cancer. Thymidylate kinase (TMK) is also involved in activation of the acquired immunodeficiency syndrome (AIDS) prodrug azidothymidine (AZT)Citation75.
Wolfenden developed a series of inhibitors of TK and TMK based on Lienhard’s finding, using a polyphosphate bridge to link adenosine and thymidine as a bisubstrate (ApndT). In a first study, Wolfenden evaluated these molecules as inhibitors of TK, trying to increase the affinity to either cytosolic enzyme or mitochondrial enzymeCitation76. The polyphosphate bridge featuring five phosphate groups was required for the greatest inhibition (11% and 18% of activity at 1.0 μM), which provided information on the distance between both binding sites. Wolfenden also reported the biological evaluation of Ap4dT, Ap5dT, Ap6dT, Ap4A, and Ap6A as inhibitors of TMKCitation77. Thymidylate kinase was inhibited by Ap4dT, Ap5dT, and most strongly by Ap6dT (Ki = 0.20 μM ATP, Ki = 0.18 μM TMP). This last compound showed competitive inhibition when either TMP (Km = 40 μM) or ATP (Km = 0.25 μM) was varied. Ap5dT was subsequently used by Reinstein et al. to solve the structure of TMKCitation75.
4.4.4. Deoxynucleoside kinases
This family consists of various deoxynucleoside kinases including cytidine (EC 2.7.1.74), guanosine (EC 2.7.1.113), adenosine (EC 2.7.1.76), and thymidine kinase (EC 2.7.1.21), which also phosphorylates deoxyuridine and deoxycytosine. These enzymes catalyze the production of deoxynucleotide 5′-monophosphate from a deoxynucleoside, using ATP as co-substrate.
Ives et al. used Lienhard and Wolfenden’s proposal to evaluate a series of multisubstrate adducts as inhibitors of deoxynucleoside kinases. The molecules were structurally similar to the one being studied for TK, TMK, and AK, but one of two adenosines in the multisubstrate was deoxygenated on the 3′-position (dApnA). dAp3A had the exact feature of the substrates for dAdo kinase. Unfortunately, it was found to be a weak inhibitor of dAdo kinase I, with a calculated Kiapp of 28 μM against ATP at a fixed dAdo concentration of 0.01 mM. In contrast, dAp5A, with one more phosphate group, appeared to be a potent and specific inhibitor for dAdo kinases, with a Kiapp of 2.7 μM for dAdo kinase ICitation78.
4.4.5. Creatine kinase
Creatine kinase (CK; EC 2.7.3.2) catalyzes the synthesis of phosphocreatine (PCr), which is subsequently used in the regeneration of ATP in cell types where the consumption of ATP is rapidCitation79. Steghens et al. reported the first synthesis and evaluation of bisubstrate inhibitors of CK combining creatine analog and hydrophobic moieties ()Citation80.
Scheme 17. Bisubstrates for creatine kinase.
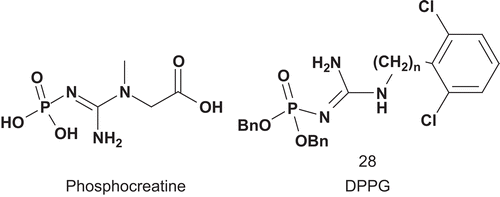
The evaluation was carried out at pH 6.6 and 8.6 for the two isoforms of CK, CK-MM and CK-BB. It appeared that at pH 6.6 there was competitive inhibition for CK-MM with DPPG versus both substrates CP and ADP, with Kiapp values of 28 μM and 1.3 μM, respectively. For CK-BB non-competitive inhibition was observed at this pH. However, at pH 8.6, competitive inhibition was detected for both enzymes CK-BB and CK-MMCitation80.
4.4.6. Phosphoglycerate kinase
Phosphoglycerate kinase (PGK; EC 2.7.2.3) is a transferase which catalyzes the interconversion of 1,3-bis-phosphoglycerate (1,3-BPG) and adenosine diphosphate with 3-phosphoglycerate (3-PGA) and adenosine triphosphateCitation81. According to Blackburn, ideal analogs should feature five negative charges in the active site in addition to two on the glycerate 3-phosphate ester, and with a linker stable to hydrolysisCitation81. Therefore, Blackburn et al. developed a chemical synthesis and determined binding characteristics for several non-hydrolyzable bisphosphonate analogs of 1,3-BPGCitation81–83. The best ligand of the enzyme was compound 29, with a Kd value of about 1 μM ().
Scheme 18. Ligand of phosphoglycerate kinase.
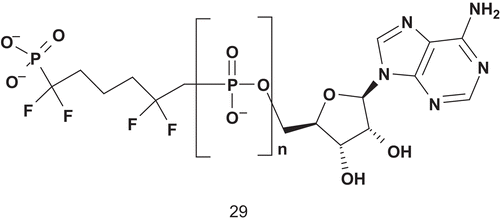
A decrease of the binding activity was also observed when the polyphosphate linker was composed of two phosphonates. This suggested that the affinity of bisubstrate analogs for PGK is dominated by coulombic interaction of the phosphoryl moietyCitation81.
4.4.7. 6-Hydroxymethyl-7,8-dihydropterin pyrophosphokinase
6-Hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK; EC 2.7.6.3) catalyzes the transfer of pyrophosphate from ATP to 6-hydroxymethyl-7,8-dihydropterin (HP), leading to the biosynthesis of folate cofactors. This is the first reaction in the folate biosynthetic pathwayCitation84. In contrast with mammals, microorganisms synthesize folates de novo, and therefore, as with all other enzymes in the folate pathway, HPPK is an interesting target for the development of an antimicrobial agent. Yan et al. synthesized three bisubstrate analogs each containing a pterin ring, an adenosine moiety, and a polyphosphate bridge as linker ()Citation85.
Scheme 19. Bisubstrate analogs for 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK).
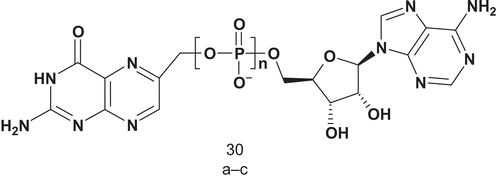
These three bisubstrates were biologically evaluated as potential inhibitors of HPPK, and it was shown that bisubstrate 30c (n = 4, IC50 = 0.44 μM) was the most potent inhibitor. The authors subsequently used 30c to determine the crystal structure of HPPK.
4.4.8. Protein kinases A, C, and IRK
Activators of protein kinase C (PKC; EC 2.7.11.13) such as phospholipids or diacylglycerol interact with the regulator domain, while both ATP and the protein substrate interact with the catalytic domainCitation86.
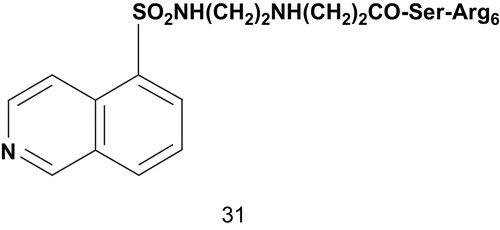
Sergheraert et al. reported the design of new bisubstrates as inhibitors of PKC which interact simultaneously with both regulator and catalytic domainsCitation87. These bisubstrates featured a cluster of arginine residues as substrate mimics, [(dimethyl-amino)naphthalenyl]sufonyl (dansyl) and 5-iso-quinolinylsulfonyl groups as ATP mimics, and the spacer was constituted of four β-alanine residues. The inhibitory activities were measured on PKC and PKA (cAMP-dependent PK; EC 2.7.11.1) with histone as substrate. The most potent inhibitor was compound 31 (), with Ki values of 0.1 μM and 0.004 μM against PKC and PKA, respectively. Compound 31 was about 60-fold more active than 1-(5-isoquinoline sulfonyl)-2-methylpiperazine toward PKC and 750-fold more toward PKACitation88.
Scheme 20. Bisubstrate analogs for protein kinase C.
Based on the previous studies, Cole et al. reported the effect of introducing an acetyl spacerCitation89. In designing the bisubstrates, they selected aminoalanine as the serine mimic linked to ATPγS via an acetyl group, with kemptide used as peptide substrateCitation90,Citation91.
The evaluation was performed on the recombinant PKA expressed from E. coli, and it was found that 32 was a competitive inhibitor versus ATP (Km = 14.1 μM), with a Ki value of 3.8 μM ().
Scheme 21. Bisubstrate analogs for protein kinase (1).
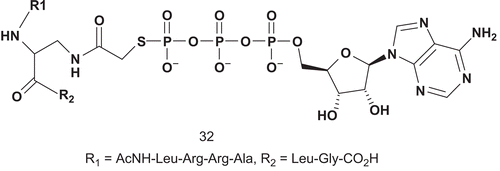
The bisubstrate analog described here was found to be more than 20-fold selective versus the related protein kinase, protein kinase C, and the less similar protein tyrosine kinase, Csk. This information highlighted the ability of the peptide moiety to contribute toward increased affinity and specificity of inhibitionCitation89.
Cole et al. also investigated a new approach in bisubstrate designCitation92. They based their study in part on the mechanism and structural consideration of the predicted dissociative transition state for PK, whereby they replaced the oxygen of tyrosine with an amino group that could be used as a hydrogen bond donor ().
Scheme 22. Bisubstrate analogs for protein kinase (2).
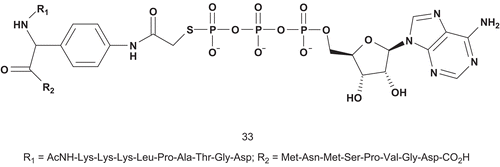
Kinase assays with compound 33 revealed it to be a potent inhibitor of insulin-receptor kinase (IRK). It was a competitive inhibitor with regard to both nucleotide and substrate, with a Ki value of 370 nM, which was 190–760-fold lower than Km values of the substratesCitation92.
4.5. Purine nucleoside phosphorylase
Purine nucleoside phosphorylase (PNPase; EC 2.4.2.1) catalyzes the reversible phosphorolysis of purine nucleosides such as inosine, 2′-deoxyinosine, guanosine, and 2′-deoxyguanosine to the purine and α-ribose or 2-deoxy-α-ribose 1-phosphateCitation93.
Inhibitors of human and parasitic PNPase are considered potential immunodepressive and anti-parasitic agentsCitation94. They may also have utility in the treatment of human T-cell leukemia, autoimmune disorders, and in the prevention of transplant rejectionCitation95,Citation96.
The diphosphate derivative of acyclovir (35) was a very potent inhibitor of the human enzyme (Ki = 8.7 nM, inosine Km = 40 μM, 2′-deoxyinosine Km = 65 μM, guanosine Km = 46 μM)Citation97,Citation98. It is a metabolically stable “multisubstrate” acyclic nucleotide analog containing a purine and a phosphate-like moiety such as 9-phosphonoalkyl derivative. The most potent inhibitor of human erythrocytic PNPase in this series was 9-(5-phosphonopentyl)guanine (34a), but its Ki value was only 170 nMCitation99. Later, Danzin reported the synthesis and the biological evaluation () of the fluoro derivative 34b (9-(5,5-difluoro-5-phosphonopentyl)guanine), the analog of the phosphonate 34aCitation.
Table 5. Comparison of inhibitor constants of 34a and 34b for purine nucleoside phosphorylase (PNPase) from various sources.
These data were the first evidence of the superiority of a difluorophosphonate compound (34b) over a phosphonate (34a) as enzyme inhibitor. Later, Tuttle et al. based their strategy on Ealick’s X-ray crystallography studies to enhance the potency of 9-phosphonoalkylguanineCitation101. The hypothesis was to incorporate, into the 9-phosphonoalkylguanine, the appropriate spaced aryl substituent with affinity for the phosphate-binding site ()Citation102.
Scheme 23. Multisubstrate adduct for human erythrocyte purine nucleoside phosphorylase (PNPase) (1).
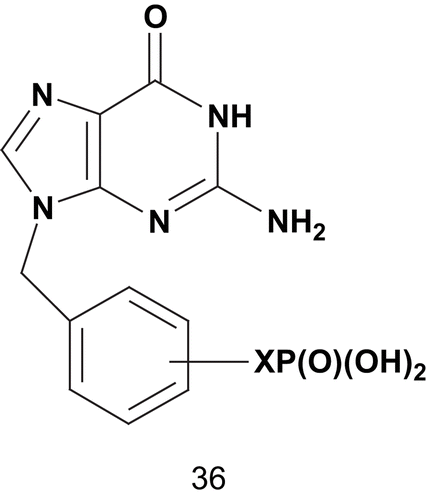
These multisubstrates 36 were tested for inhibition of human erythrocyte PNPase via a xanthine oxidase coupled assay with inosine as variable substrate. From this series, compounds 36 with X = 3-CH2-OCH2 (Ki = 5.8 nM) and X = 3-CH2SCH2 (Ki = 1.1 nM) were among the most potent inhibitors of PNPase reportedCitation102. After this study, Tuttle et al. envisaged synthesizing a stable mimic of the diphosphate 37a in which the oxygenated side chains were replaced by methylene moieties, as for (phosphinicomethyl)phosphate acid 37b ()Citation93.
Scheme 24. Multisubstrate adducts for human erythrocyte PNPase (2).
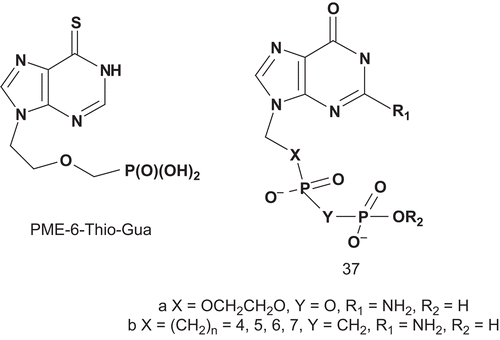
The compounds were tested for inhibition of human erythrocyte PNPase. Two new potent inhibitors 37b, n = 5 and n = 6, which were stable mimics of 37a, were identified. They had Ki (inosine as variable substrate) in the nanomolar range when assayed in the presence of zinc chlorideCitation94. From this study, a model assuming interacting binding sites was more probable than the hypothesized dependent sites model.
4.6. Methyl transferases
4.6.1. Catechol O-methyltransferase
Many enzymes catalyze transmethylation using S-adenosylmethionine (AdoMet or SAM), and catechol O-methyltransferase (COMT; EC 2.1.1.6) is one of them. Inhibition of COMT is considered an important approach in the development of new therapeutic treatments of Parkinson’s disease, and recently this enzyme has been implicated in the modulation of painCitation103,Citation104.
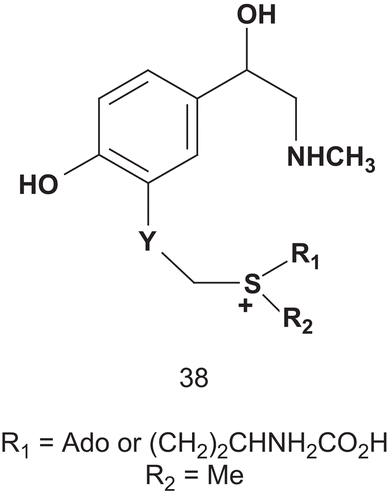
In a first study, Coward reported the synthesis and evaluation of novel potential multisubstrate inhibitors of COMTCitation105. The synthesis was directed toward compound 38, which was assumed to be a potential transition-state analog.
Therefore, a series of compounds, 38, analogs of 34 and incorporating either a homocysteine or an adenosyl moiety and a sulfonium center, were synthesized (), and were shown to have satisfactory inhibitory activities with a Ki of approximately 1 mMCitation106. Coward then designed other potential multisubstrate inhibitors of COMT and phenethanolamine N-methyltransferase (PNMT; EC 2.1.128). Unfortunately, the compounds had only weak inhibitory activity against COMT, and none of them were inhibitors of PNMT in assays using β-phenethanol-amine and AdoMet as substrates. Even though two inhibitors of COMT have now reached the market, there is to date no lead structure for bisubstrate inhibitors of this enzymeCitation107. Therefore, Diederich et al. described a rational design, synthesis, and evaluation of new bisubstrate inhibitors of COMT ()Citation108. They based their research on determination of the crystal structure of COMT complexed with SAM, 3,5-dinitrocatechol, and magnesium ionCitation109.
Scheme 25. Multisubstrate for catechol O-methyltransferase (COMT) (1).
Scheme 26. Multisubstrates for COMT (2).
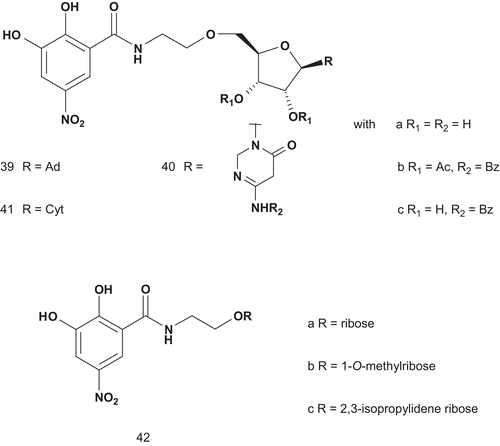
These compounds were tested as inhibitors of COMT isolated from rat liver. The results showed 39a as the most potent inhibitor (IC50 = 2 μM with preincubation, 4 μM without preincubation) as it was 10-fold more potent than 42b (IC50 = 25 μM with preincubation, 26 μM without preincubation). This behavior could be explained by the nature of the substrate, which allowed 39a to reach both catechol and SAM pockets. Compounds 42a (IC50 = 26 μM with preincubation, 25 μM without preincubation) and 42b (IC50 = 35 μM with preincubation, 27 μM without preincubation) had similar inhibitory activities, indicating that addition of the ribose component did not improve on the inhibitory effect of the catechol moietyCitation108.
Only recently has the first effective bisubstrate COMT inhibitor been developed, and for this type of compound they used both the nucleoside and catechol moieties to bind the SAM and substrate enzyme binding sitesCitation110,Citation111. This work was extended by synthesizing a new series of bisubstrate inhibitors reaching binding activities in the nanomolar range ()Citation112.
Table 6. IC50 values for the bisubstrate inhibitors 43 and 44 of catechol O-methyltransferase (COMT).
Thus, Diederich et al. showed that affinity for COMT strongly depends on the size and shape of the linker between the nucleoside and catechol moieties. Adverse effects of hepatotoxicity have been associated with the use of tolcapone, and the hypothesis is that hepatotoxicity might be correlated to the nitrocatechol groupCitation113,Citation114. Thus, Diederich et al. focused their efforts on the development of a new generation of potent bisubstrate inhibitors of COMT lacking the nitro group. Previously, they reported a potent bisubstrate inhibitor (44a, IC50 = 9 nM), and by crystal-structure analysis and kinetic study they demonstrated that 44a bound both the SAM and the substrate binding sites of COMTCitation112. Therefore, based on this result they modified 44a by replacing the nitro group by a hydrophobic groupCitation115. This study provided the first family of bisubstrate inhibitors of COMT with an IC50 in the nanomolar range. In addition, this demonstrated that the nitro group was not required for a high binding activityCitation112.
4.6.2. Methionine S-adenosyltransferase isozyme
Methionine S-adenosyltransferase (MAT; EC: 2.5.1.6) is an enzyme which catalyzes the attack of the sulfur atom of l-methionine on C5′ of ATPCitation116–118. Hampton et al. reported the synthesis of new potential inhibitors for two isozymes of MAT: M-2 and M-TCitation116–118. A preliminary study looked at the effect of the distance between S and C5′ in the covalent adduct 45 of l-methionine and β,γ-imido-ATPCitation116–118. Compounds were tested as inhibitors of M-2 and M-T forms of methionine S-adenosyltransferase ().
Table 7. Inhibition constants of adenine nucleotide derivatives on M-2 and M-T.
In contrast with the previous polyphosphate compounds, the incorporation of a phosphoramidate increased the activity 2.5-fold, with improved metabolic stability. These results showed that compound 45 was the most potent inhibitor for both isozymes M-2 and M-T. The carbon chain was increased in 45–46, which slightly decreased the inhibitory activity. Ribose-P• elongation by one methylene carbon between C4•-C5•, and also between C5•-C6•Citation.
The results showed that this elongation was compatible with the maintenance of dual-site inhibition activity, but did not exhibit more selectivity. It was also found that replacement of the 6-amino group of the ATP-moiety by S-nBu gave a compound three-fold more selective for the target isozyme. Hampton reported a new series of potential inhibitors which were evaluated and compared for potency with 45Citation116–118,Citation120.These compounds showed an inhibitory activity comparable to 45 but were found to have great selectivity between M-2 and M-T when methionine was used as substrate.
4.6.3. Indole N-methyltransferase
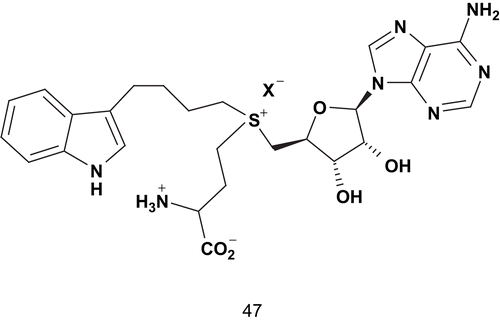
Indole N-methyltransferase (INMT) is an S-adenosylmethionine (SAM)-dependent enzyme. It catalyzes the conversion of tryptamine into N-methyltryptamine (NMT) and N,N-dimethyltryptamine (DMT)Citation121. Crooks reported the design of a selective in vivo inhibitor of tryptamine N-methylation which did not disturb other SAM-dependent methylation reactions. This study aimed to determine the role of the tryptamine N-methylation in psychotic disorders based on the definition of the COMT transition state. Crooks proposed a similar structure () for the INMT reaction, with tryptamine as substrateCitation121. This structure, 47, represented a stable synthetic analog of this transition state in which the side-chain amino group of tryptamine and the methyl, which migrates, were replaced by a saturated carbon chain.
Scheme 27. Synthetic transition-state analog of indole N-methyltransferase (INMT).
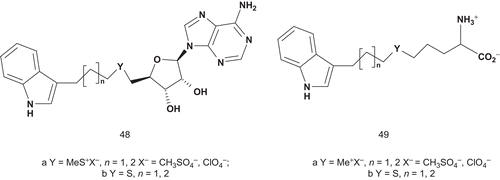
Therefore, the synthesis was directed toward the thioether precursors of 47 and their analogs, methylsulfonium salts (). These compounds were evaluated as inhibitors of INMT, and the most potent inhibitor appeared to be 48a (Y = MeS+X−, n = 2, X− = ClO4−, IC50 = 38 μM). The lack of inhibitory activity of thioether structures 49b (Y = S, n = 1, 2) and 48b (Y = S, n = 1, 2) suggested that a positive charge borne by the sulfur atom was required for INMT binding.
Scheme 28. Multisubstrates for INMT.
4.7. Thymidine phosphorylase
Thymidine phosphorylase (TPase; EC 2.4.2.4) has been implicated in angiogenesis and chemotaxis in human tumorsCitation122,Citation123. Over-expression of TPase has also been implicated in inflammatory disease states including rheumatoid arthritis and psoriasisCitation124,Citation125. TPase catalyzes the reversible phosphorolysis of thymidine to thymine and 2-deoxyribose 1-phosphateCitation126. Balzarini et al. reported the first series of MAIs for TPase () in order to obtain detailed enzymatic kinetics of the bacterial TPase and identification of two different enzyme binding sitesCitation127.
Scheme 29. Multisubstrates for thymidine phosphorylase.
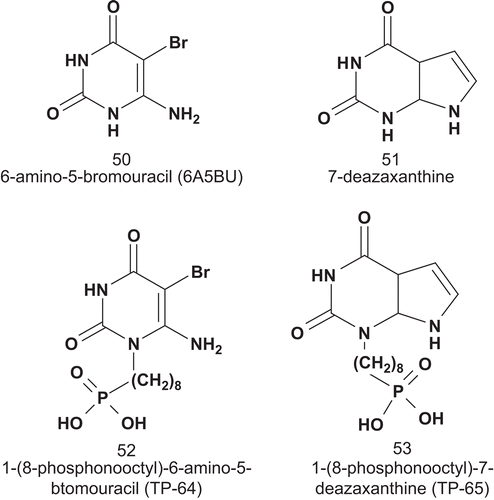
The inhibition kinetics of these compounds against bacterial TPase showed clearly that 52 and 53 inhibited TPase in a competitive or mixed fashion. The Ki values were 3.2, 2.1, 476, and 125 μM for 50, 51, 52, and 53, respectively.
Recently, Allan et al. described a novel series of multisubstrate analogs based on compound 55a, synthesized by Li and GamenCitation128,Citation129. These compounds were evaluated as inhibitors of human recombinant TPase (). Unfortunately, the replacement of the phosphonate group with a carboxylic acid (54b) reduced the inhibitory activity.
Table 8. Inhibition of thymidine phosphorylase (TPase) by multisubstrate analogs.
The substitution to 54c rendered the compound inactive. For both types of analog, the endo-isomer was 30–60 times more active than the exo-compound but less active than 54aCitation128.
4.8. Glycosyltransferases
4.8.1. α-1,2-Fucosyltransferase
α-1,2-Fucosyltransferase is known to transfer a fucosyl residue from guanosine 5′-diphosphofucose (GDP-fucose) to the 1-OH group of β-d-galactopyranosides with inversion of configuration at the fucopranosyl anomeric carbon. Hindsgaul et al. reported the first specific glycosyltransferase inhibitors using the mechanism-based approach () to design MAIsCitation130.
Scheme 30. Multisubstrates for α-1,2-fucosyltransferase.
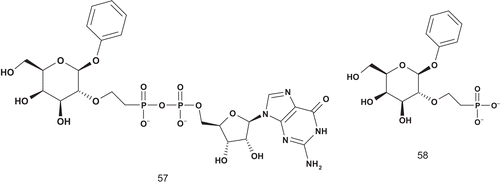
First, the bisubstrate 57 was kinetically evaluated using the membrane-bound form of fucosyltransferase. The mode of inhibition was competitive with regard to both GDP-fucose (Km = 7.3 μM) and β-phenyl galactopyranoside (acceptor, Km = 2600 μM), with Ki values of 16 μM and 2.3 μM, respectively. In contrast, Ki values of GDP-fucose and β-phenyl galactopyranoside were 7.3 μM and 2.6 μM, respectively. These data proved that inhibitor 57 could occupy both the GDP and acceptor binding sites as required for a true bisubstrate analogCitation130.
Second, further experiments were performed to evaluate the contribution of GDP in inhibitory activity. It was found that 58 (phosphonate) was a competitive inhibitor with respect to the acceptor and a mixed inhibitor with regard to GDP-fucoseCitation130. It was concluded that the binding properties of 57 were strongly dependent on recognition of the GDP moiety, since the Ki of 58 was increased over 50-fold compared with 57. The bisubstrate analog was also found to be an effective inhibitor of the soluble form of α-1,2-fucosyltransferase.
4.8.2. β-1,4-Galactosyltransferase
Hashimoto et al. reported the synthesis of a multisubtrate inhibitor of glycotransferase and more particularly β-1,4-galactosyltransferase (GlcNAc; EC 2.4.1.22)Citation131. This multisubtrate was composed of three components: an electrophilic glycosyl residue (Gal), a nucleotide-leaving group (UDP), and a nucleophilic glycosyl acceptor (GlcNAcβ-OMe) ().
Scheme 31. Multisubstrate for β-1,4-galactosyltransferase.
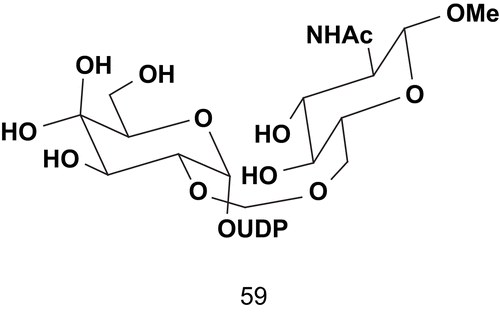
This multisubstrate 59 adduct, the design of which was based on a hypothetical mechanism, showed a remarkably potent inhibitory activity toward β-1,4-galactosyltransferase from bovine milk (Ki = 1.35 μM for acceptor GlcNAc and Ki = 3.3 μM for donor UDP-Gal)Citation131,Citation132. In addition, this compound was the first multisubstrate example that showed inhibitory activity against GlcNAc.
Later, Guillerma et al. reported the synthesis of a bisubstrate analog prototype featuring a five-membered ring azasugar with l-xylose stereochemistry, in order to mimic the half chair conformation of the glycosyl cation. A difluoromethylphosphonate group was incorporated at the C1 position to resemble the first position of the pyrophosphate moiety of the donor. They also introduced an amino group in the linker between the azasugar and GlcNAc, expecting an additional electrostatic interaction between the 4-position of the acceptor and the enzymeCitation132. This bisubstrate was tested as an inhibitor for chitin synthase (an important target for antifungal agents), but showed no activity.
4.8.3. Sialyltransferase
Sialyltransferases (STs; EC 2.4.99.2) are involved in biosynthesis of the sialic acid-containing oligosaccharidesCitation133. Most STs use cytidine-5′-monophospho-N-acetylneuraminic acid (CMP-Neu5Ac) as common donor substrate. Several stable donor analogs of glycosyltransferase were synthesized, and Schmidt et al. reported the most potent inhibitor with a Ki value of 29 nMCitation134–141. The same group also synthesized several multisubstrate adducts, but none had inhibitory effect. Hinou et al. synthesized analogs with Ki values similar to the Km value of CMP-Neu5AcCitation142. Based on a similar idea, Hashimoto et al. reported the synthesis of donor analogs 60a–d of CMP-Neu5Ac, the multisubstrate adduct 60e (), and their evaluation as inhibitors of α-2,3-ST and α-2,6-ST ()Citation143.
Table 9. Inhibitory activities of 61–65 against rat recombinant α-2,3-sialyltransferase (ST) and α-2,6-ST.
Scheme 32. Multisubstrates for sialyltransferases.
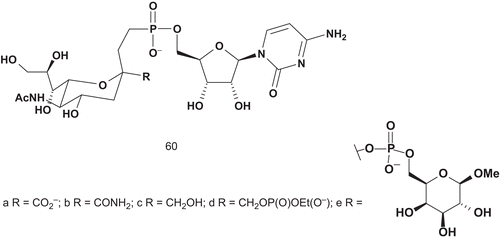
Only the carboxylate derivative showed interesting inhibitory activities against both STs (IC50 = 0.047 mM for α-2,3-ST and 0.34 mM for α-2,6-ST). The difference between these two IC50 values was about one order of magnitude, so compound 61a was postulated to be a good lead structure for the design of selective bisubstrate analog inhibitors.
4.8.4. α-1,3-Fucosyltransferase
After having developed bisubstrate inhibitors of β-1,4-galactosyltransferase, Hashimoto used the same concept to synthesize bisubstrate analogs targeting α-1,3-fucosyltransferaseCitation144. Two compounds were designed as bisubstrate analogs for α-1,3-fucosyltransferase, and their inhibitory activities were determined against both α-1,3-fucosyltransferases (α-1,3-FucT) V and VI ().
Table 10. Inhibitory activities of bisubstrate analogs 66a and 66b.
These compounds were found to be weak inhibitors for FucT-V but substrates of FucT-VI. This discovery provided new insight into the substrate binding site of these two gene products, and useful information for the development of FucT-V-specific inhibitors. It also helped establish the utility of FucT-VI in order to modify fucosylated glycoconjugatesCitation144.
4.8.5. N-acetylglucosaminyltransferase
N-Acetylglucosaminyltransferases (GnTs; EC 2.4.2.51) are key enzymes in the production of branched complex N-glycan structures. GnTs transfer an N-acetylglucosamine (GlcNAc) residue to the core α-1,6-mannose (Man) to form a β-1,6 linkageCitation145,Citation146. GnT-V affects T-cell activation and angiogenesisCitation147–152. Therefore, inhibition of GnT-V might have potential in the treatment of cancer. Several GnT-V inhibitors were synthesized based on modification of the acceptor substrate oligosaccharide.
Yukishige et al. turned their attention to designing multisubstrates relying on the incorporation of a donor into an acceptor, expecting to enhance binding and inhibitory activityCitation153. They reported the synthesis of a prototype for bisubstrate-type inhibitors of GnT-V and GnT-IX (). This type of bisubstrate was designed to contain both donor (UDP-GlcNAc) and acceptor componentsCitation154. As the acceptor component, the trisaccharide (GlcNAcb-1,6-Man) was incorporated, as it had previously been reported to serve as an efficient acceptor substrate of GnT-VCitation155.
Scheme 33. Bisubstrate for N-acetylglucosaminyltransferases GnT-V and GnT-IX.
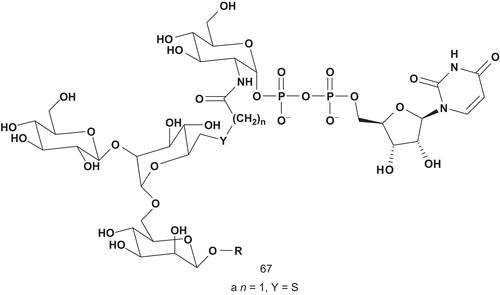
This compound 67a was evaluated toward GnT-V and GnT-IX (). The affinity of 67a to GnT-V was weak compared with the acceptor substrate (Km = 150 μM). However, the activity toward GnT-IX was more important, so much so that 67a could be used as a probe to investigate the kinetic mechanism of GnT-IX.
Table 11. Inhibitory activities of 67 against N-acetylglucosaminotransferases GnT-V and GnT-IX.
The same group developed several analogs of 67a. They studied the dependence of the length of the linker Y () and evaluated these compounds against GnT-V and GnT-IX ()Citation153. The results showed that inhibition was clearly dependent on the length of the linker. For GnT-V, compounds which had the longest linker showed the strongest activity, with a single exception, 67a.
The correlation between linker length and activity was significantly different between these enzymes. Although these enzymes are homologous, the biological results suggested that the distances for donor and acceptor binding sites were quite different.
4.9. Acetyltransferases
4.9.1. Serotonin N-acetyltransferase
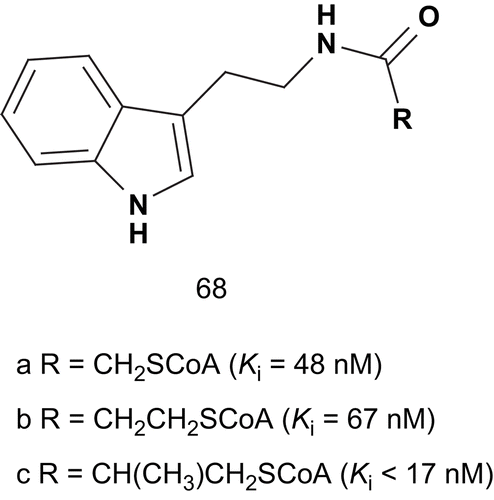
Serotonin N-acetyltransferase, also known as arylalkylamine-N-acetyltransferase (AANAT; EC 2.3.1.87), catalyzes the transfer of an acetyl group from acetyl coenzyme A (AcCoA) to the primary amine of serotonin to give N-acetylserotonin. AANAT catalyzes this transfer via a ternary complex kinetic mechanism; therefore, it is relevant to assume that bisubstrate analog inhibitors composed of tryptamine linked with CoA might result in potent inhibitionCitation156,Citation157. Bisubstrate analog inhibitors have been used along with AANAT to solve the X-ray structure of the AANAT–bisubstrate complex, which has improved understanding of molecular recognition and the catalytic mechanism.
Cole et al. directed their studies toward the design of bisubstrates in which tryptamine was linked to CoA via an acetyl bridgeCitation158. This compound displayed a Ki value about 1000-fold lower than the substrate Km value (68a). Many studies developed several bisubstrate analogs with substitution in the indole, CoA, and linker moieties, and these were evaluated as AANAT inhibitors. Studies revealed that the methylene extension of the linker (68b) led to inhibitors as potent as 68a. They also demonstrated that the attachment of an extra methylene group in the linker (68c) led to even more potent inhibition ().
Scheme 34. Bisubstrates for serotonin N-acetyltransferase (1).
Later, the same group concentrated its attention on preparing a series of novel bisubstrate ketone analogs, deaza analogs of 68b, hoping to gain further insight into the role of the linking region in AANAT inhibitionCitation156. One of these analogs was shown to have an apparent Ki about two-fold less than that of the parent compound 68aCitation159. This demonstrated that the amide nitrogen of 68a was not important for a high affinity interaction with AANAT. The study of these bisubstrate ketone analogs revealed that the tetrahedral intermediate bisubstrate mimic only weakly blocked AANAT action.
In addition, the relative orientation of the two substrate moieties with respect to each other appeared to be important for potent inhibition. With the aim of potentially enhancing inhibition and gaining new mechanistic insights, Cole et al. reported further modifications of previously synthesized bisubstrates ()Citation156,Citation160. These AANAT bisubstrate analog inhibitors allowed for hydrogen binding and electrostatic interaction in molecular recognition sites. Analogs 69a–c were designed to investigate the dependence of the inhibitory potency on the distance between the indole, which was kept intact due to its mimicry of tryptamine, and CoASH moiety in AANAT bisubstrate analogs.
Scheme 35. Bisubstrates for serotonin N-acetyltransferase (2).
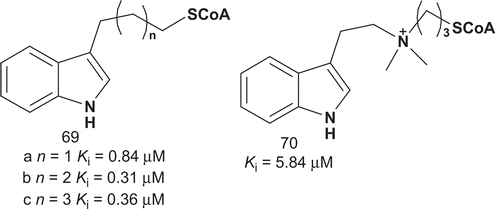
Bisubstrates 69a–c were evaluated as inhibitors of AANAT (), and these results suggested that the linker’s ability to undergo hydrogen-bond interactions made only modest contribution to the enzyme–adduct complex’s stability. In addition, the linker between the indole and CoASH moieties required at least four methylenes to allow for strong AANAT inhibition. Compound 70 evaluation showed that the AANAT active site was not adapted to support a positively charged linker.
4.9.2. Histone acetyltransferases
Histone acetyltransferases (HATs; EC 2.3.1.48) are enzymes which acetylate conserved lysine amino acids on histone proteins by transferring an acetyl group from acetyl CoA to lysine to form ϵ-N-acetyl lysine. Cofactors p300 and CBP (CREB binding protein) have been shown to be major regulators of gene expression via their HAT functionCitation161. In addition, p300/CBP HAT activity appears to be enhanced in some types of cancer and, thus, selective p300/CBP HAT inhibitors may have utility as therapeutic agentsCitation162–164.
Cole et al. reported several studies of the structure and substrate processing, before designing bisubstrate analog inhibitors ()Citation164–168. These bisubstrates were analogs with Lys-CoA, and studies of the substructures of the CoA moiety of Lys-CoA revealed that the CoA, without modification, was crucialCitation169.
Scheme 36. Bisubstrate analogs 71 and 72 for histone acetyltransferases.
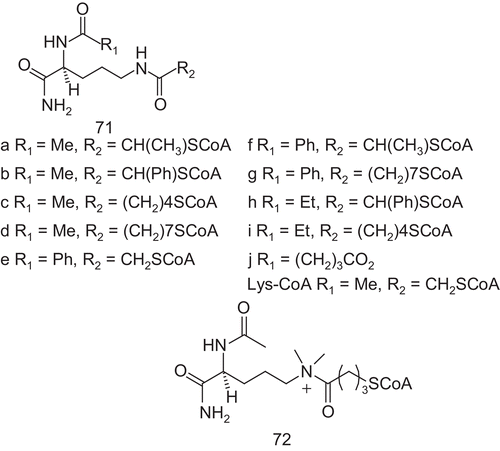
The Lys-CoA analogs were tested against the catalytic domain of p300 and they were referenced to Lys-CoA. Most compounds showed IC50 values similar to or worse than those of Lys-CoA. Modification of the linker with phenyl substitution or homologation of the alkyl chain reduced the inhibitory activities. In addition, deletion of the carbonyl group from the linker decreased the inhibitory potency. Compared to Lys-CoA, only 71a and 71e enhanced the potency with IC50 values of 0.8 μM and 0.7 μM respectively, about four-fold lower than Lys-CoA. In contrast, the double modification operated with 71f showed a similarly potent inhibition to that of Lys-CoA, indicating a cancelation of the affinity-enhancing effects of each substitution.
4.9.3. Aminoglycoside 6′-N-acetyltransferase (AAC(6′)-li)
Aminoglycosides are efficient antibiotics and are particularly active against aerobic gram-negative bacteriaCitation170. Nevertheless, the emergence of aminoglycoside resistance has strongly restricted their use as antibacterial agentsCitation171. Auclair et al. developed a regio- and chemoselective methodology for direct N-6′-derivatization of unprotected aminoglucosides, facilitating the preparation of a first series of AAC(6′)-li bisubstrate analog inhibitors ()Citation172,Citation173.
Scheme 37. Bisubstrate analogs for aminoglycoside 6′-N-acetyltransferases (1).
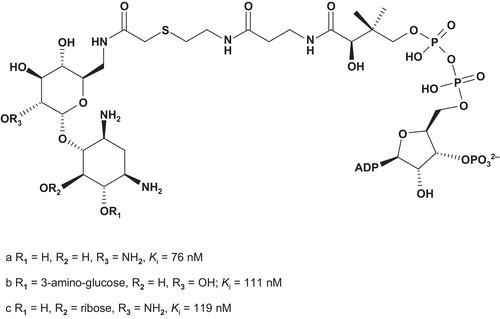
These target bisubstrates were designed according to the proposed tetrahedral intermediate that resulted from the attack of aminoglycoside 6′-NH2 on the thioester carbonyl of AcCoA in the active site of the enzymeCitation174. Kinetic studies of 73a–c showed these bisubstrates as potent competitive inhibitors, with nanomolar Ki values. Based on these results, Auclair synthesized a novel series of bisubstrates () to investigate the importance of the size and geometry of the linkerCitation156–160,Citation173.
Scheme 38. Bisubstrate analogs for aminoglycoside 6′-N-acetyltransferases (2).
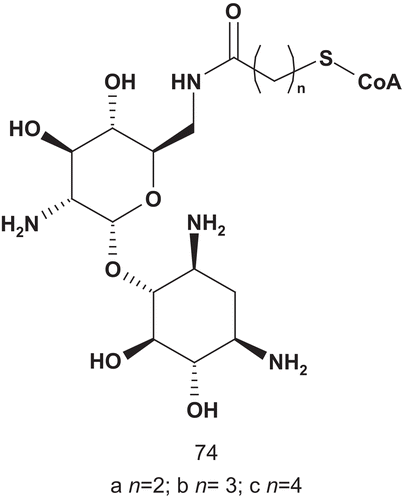
AAC(6′)-li inhibition assays showed that 74a was the most potent bisubstrate inhibitor of this series. The enzyme bound 74a about 200-fold tighter than its natural substrate AcCoA (Km = 9.6 μM). It was also observed that an increase in length of the linker led rapidly to a decrease in activity.
The high potency of these bisubstrate inhibitors allowed crystallization of AAC(6′)-li with aminoglycoside derivatives that provided valuable guidance in further studies of this enzyme. Recently, the same group reported a novel series of bisubstrate inhibitors of AAC(6′)-li in order to carry out a structure–activity relationship studyCitation175. The inhibitory activity of this new generation of bisubstrate analogs was evaluated with AAC(6′)-li, but none had a low, nanomolar Ki value. The SAR from these studies indicated that inhibition by aminoglycoside-CoA bisubstrates was more sensitive to truncation at the aminoglycoside than at the CoA end.
Targeting the same family of enzymes, Mobashery et al. reported the development of tethered bisubstrates ()Citation176. They focused their attention on aminoglycoside 3′-phosphotransferases (APH3′; EC 2.7.1.95), which catalyze the transfer of the γ-phosphoryl group of ATP to the 3′-hydroxyl of aminoglycosides. APH3′ enzymes were the cause of the demise of kanamycin treatment in clinical trials.
Scheme 39. Bisubstrate analogs for aminoglycoside 3′-phosphotransferases.
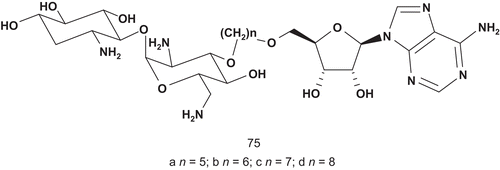
Compounds 75a–d were tested against APH3′ Ia and IIa as potential inhibitors. All of them were competitive inhibitors for the enzyme, and 75 showed the best inhibitory activity with a Ki value of 3 μM and 9 μM, respectively, when the substrate of the enzyme was kanamycin A (Km = 4 μM).
4.9.4. Aspartate transcarbamylase
Aspartate transcarbamylase (ATCase; EC 2.1.3.2) catalyzes the first unique step of the pyrimidine pathway, which is carbamylation of the amino group of l-aspartate by carbamylphosphate to produce N-carbamyl-l-aspartateCitation177. This enzyme is an interesting target for the development of antiproliferative drugs, and numerous approaches to the design of new antitumoral agents were based on the search for ATCase inhibitorsCitation178,Citation179. So far, the most potent synthesized inhibitor is the bisubstrate analog, N-(phosphonoacetyl)-l-aspartate (PALA)Citation180,Citation181. PALA is a competitive inhibitor in respect to carbamyl phosphate, but not with l-aspartate.
The application of this bisubstrate has facilitated studies about the regulation, binding characteristics, and crystallographic structure of ATCaseCitation182–186. More recently, Grison et al. investigated the rational design of ATCase inhibitors based on the understanding of the catalytic reaction mechanismCitation187. They synthesized several bisubstrate analogs, one transition-state analog, and competitive reaction inhibitors ().
Scheme 40. Bisubstrates, transition-state analog, and competitive inhibitors for aspartate transcarbamylase (ATCase).
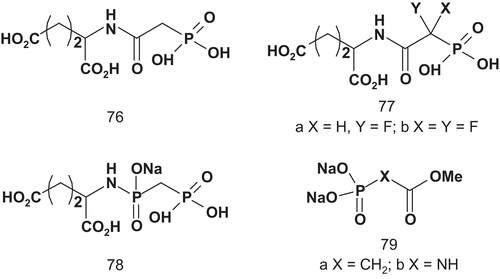
The influence of these compounds was examined on ATCase activity from E. coli. Compounds 76, 78, and 77a did not show any inhibitory activity, although, at a concentration of 5 mM, 77b inhibited ATCase at about 45%. Only 79a and 79b showed interesting inhibitory activities. At a concentration of 0.05 mM, the inhibition was about 50%, and rose to 80% at 0.33 mM.
4.9.5. Sulfotransferases
Sulfotransferases (EC 2.8.2.2 [OH] or EC 2.8.2.3 [NH2]) catalyze the transfer of a sulfuryl group from 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to a hydroxyl (or amino) group on a carbohydrate, protein, or small molecule acceptorCitation188–190. It has been demonstrated that sulfotransferases have strong connections with several disease states; thus, this discovery has positioned sulfotransferases as important therapeutic targetsCitation191.
Bertozzi et al. designed a library of bisubstrate analog inhibitors. Their strategy was to create a library () possessing two elements: a PAPS mimic for recognition toward sulfotransferases, and a variable hydrophobic and drug-like component to bind the acceptor pocketCitation192.
Scheme 41. Library of bisubstrate analogs for sulfotransferases.
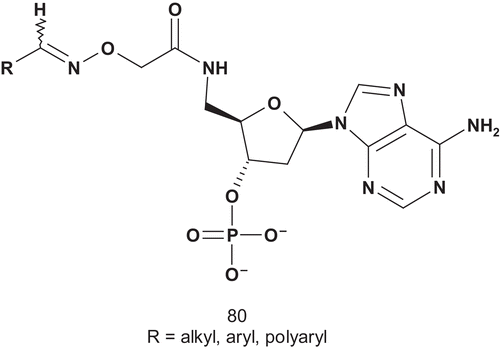
All compounds were tested as inhibitors of estrogen sulfotransferase (EST; EC 2.8.2.4), and they displayed greater than 50% inhibition at 200 μM; they also shared a greater than 55% ion abundance decrease in IEMSP (immobilized enzyme mass spectroscometry)Citation192. Other bisubstrate adducts, stemming from this study, were further developed (; R = estrone)Citation193. These compounds were also tested against EST, and were found to display moderately potent inhibitory activity. Competitive behavior was observed against PAPS (Km = 2.5 nM) with a Ki(comp) of 2.9 nM, and non-competitive behavior against substrate estrone (Km = 50 nM) with a Ki(non-comp) of 4.0 nMCitation193.
4.10. Farnesyl/geranylgeranyl transferases
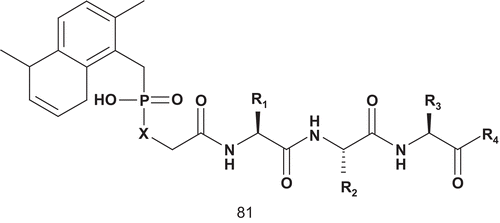
Farnesyl transferase (FPT; EC 2.5.1.58) catalyzes the transfer of a farnesyl residue from farnesylpyrophosphate (FPP) to the thiol of a cysteine side chain of proteins bearing the CAAX-tetrapeptide sequenceCitation194,Citation195. FPT catalyzes a bisubstrate reaction, and this offered many opportunities for inhibitor design. Thus, several inhibitors were prepared based on either of the two substrates, and examples of both CAAX- and FPP-based inhibitors were reported in the literatureCitation196–200. Patel et al. described the first series of potent bisubstrate analog inhibitors of FPT ()Citation201,Citation202. Their strategy in inhibitor design was to keep the farnesyl group of FPP in order to preserve putative hydrophobic interactions, and the C-terminal tripeptide was chosen as the peptide substrate componentCitation201,Citation202. These two parts were linked via a phosphonic or phosphinic acid linker to replace the sulfhydryl group.
Scheme 42. Bisubstrate analogs for farnesyl transferase (FPT) (1).
These inhibitors were evaluated for selectivity using the closely related enzyme geranylgeranyl protein transferase type I (GGT-I) ().
Table 12. Inhibitory activities of 81 against GnT-V and GnT-IX.
Compounds 81a–f were only moderately active against GGT-I (), thereby affording greater than 1000-fold selectivity in favor of the targeted enzyme FPT. In contrast, methyl prodrugs 81d and 81e showed a 75–80% decrease in transformed foci at 100 μM concentration. At 100 μM concentration both 81e and 81f almost completely suppressed colony formation of H-ras transformed NIH 3T3 cells in soft agarCitation201,Citation202. Unlike the tetrapeptide-based inhibitors, a free sulfhydryl group is not a requirement for the activity of these phosphonic and phosphinic acid bisubstrate inhibitors.
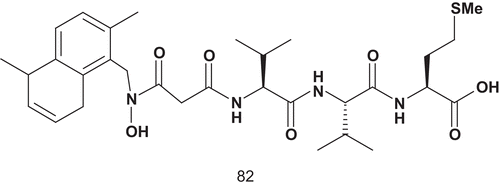
Patel et al. then developed a series of novel hydroxamic acid-based bisubstrate analog putative inhibitors of FPT. Thus, the farnesyl group and the tripeptide group of the C-terminal CAAX motif were linked together via a hydroxamic acid (R-N(OH)C(O)-R′). The introduction of a hydroxamate group as a linker instead of a branched functional group was to avoid any problems of chirality in this part of the molecule. The replacement of the sulfhydryl group found in tetrapeptide CVLS (I50 = 1 μM) by an N-methylhydroxamic acid was unsuccessfulCitation203.
Only compound 82 () was found to be a two orders of magnitude better inhibitor than the first lead, 81. Compound 82 was effective in blocking prenylation of protein in the whole cell, including p21ras.
Scheme 43. Bisubstrate analogs for FPT (2).
The presence of a free thiol in these types of molecules was a source of adverse drug effects, as observed with the angiotensin converting enzyme inhibitor captopril. Patel et al demonstrated that the free sulfhydryl group was not necessary for inhibitory activityCitation201,Citation202,Citation204. Hence, Schlitzer et al. reported a series of non-thiol farnesyltransferase inhibitors as bisubstrate inhibitors ()Citation205.
Scheme 44. Bisubstrate analogs for FPT (3).
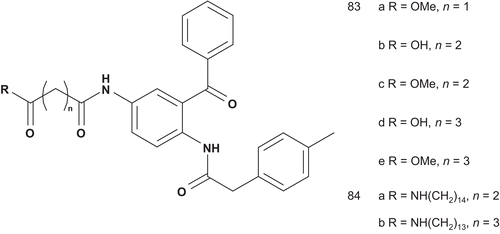
The evaluation of these bisubstrates (83a–e and 84a–b) showed very weak inhibitory activity of the farnesyltransferase, and there was no difference between a carboxylic group terminal and the corresponding methyl ester. This can be explained by the loss of binding energy upon replacement of the thiol by a carboxyl groupCitation205. Efforts then focused on development of both the peptidic and prenylic substrates and studying the structural requirements of the central moietyCitation206,Citation207.
Aromatic acrylic acids were then identified as farnesyl mimetics, and Schlitzer et al. developed a new series of bisubstrates where the aliphatic farnesyl surrogate was replaced by an aromatic acrylic acid derivative ()Citation208,Citation209.
Scheme 45. Bisubstrate analogs for FPT (4).
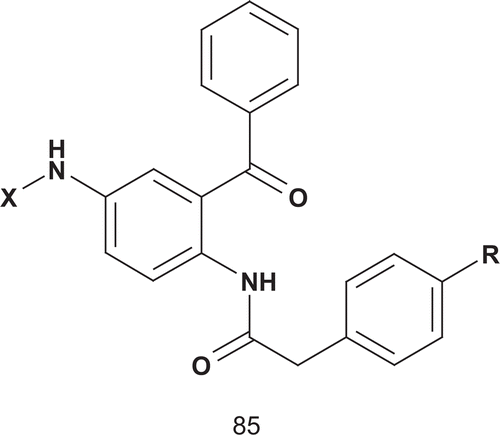
Compared to the benzophenone-based bisubstrate analog farnesyltransferase inhibitors (85a)Citation210, compounds 85b–i, bearing an aromatic farnesyl mimetic instead of an aliphatic moiety substituent, exhibited weak inhibitory activity against farnesyltransferase (). However, some of these compounds displayed concentration-dependent cytotoxicity or (at higher concentration) even a cytocidal effect against MCF-7 breast cancer cells. The compounds 85d and 85f were more active than the antitumor drug cisplatinCitation209.
Table 13. Farnesyltransferase inhibitory activity of compounds 85a–i.
Geranylgeranyl transferase-1 (GGT-1; EC 2.5.1.59) performs the same reaction as FPT but with geranylgeranylphosphate as a substrateCitation211. Substrate specificity toward FPT and GGT-1 is determined by the C-terminal residue X in the CAAX-box motif of Ras or other closely related small GTPasesCitation212. FPT preferably uses substrates where X is Met, Ser, Gln, or Ala, while GGT-1 favors substrates when X is Leu or PheCitation213,Citation214.
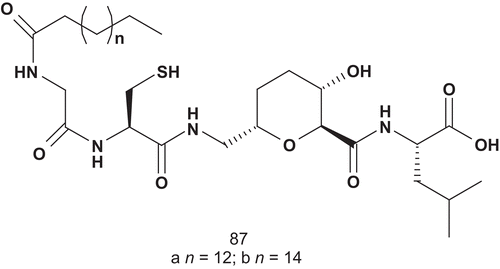
Overhand et al. developed the next generation of bisubstrate potential inhibitors of GGT-1Citation215. They had reported a series of CAAL analogs featuring sugar amino-acid based dipeptide isosteres as replacement of the central AA dipeptide, and the most potent compound () had an IC50 value of 68 μMCitation216.
Scheme 46. Bisubstrate analogs for geranylgeranyl transferase-1 (GGT-1) (1).
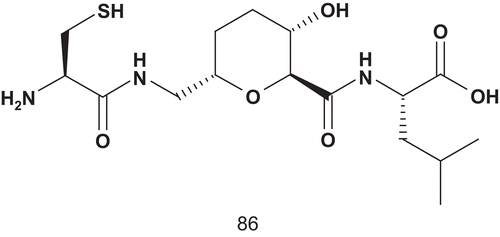
To enhance the potency, Overhand et al. attached isoprenyl moities to these CAAL analogs, leading to a bisubstrate mimicCitation215. This generation of bisubstrate analogs was evaluated as inhibitors of GGT-1, and two compounds revealed interesting inhibitory properties (). Compounds 87a (IC50 = 12.7 ± 1.3 μM) and 87b (IC50 = 12.3 ± 1.0 μM) inhibited GGT-1 with equal efficiency, although they differed in the nature of the side chain.
Scheme 47. Bisubstrate analogs for GGT-1 (2).
4.11. Riboflavin synthase
Riboflavin synthase (EC 2.5.1.9) catalyzes an unusual dismutation reaction involving the transfer of a four-carbon unit from one molecule of 6,7-dimethyl-8-d-ribityllumazine bound at the donor site of the enzyme to a second identical lumazine molecule located at the acceptor site of the enzyme. This transfer results in the formation of one molecule of riboflavin and one of pyrimidinedioneCitation217–219. Due to the fact that riboflavin synthase uses two identical molecules of lumazine, the MAI approach was used by Cushman et al. to identify potential riboflavin synthase inhibitors. They developed a series of potential bisubstrate adducts by attaching two units of 6,7-dimethyl-8-d-ribityllumazine with a carbon chain as linker. Additionally, they also examined the activity of the two units of pyrimidinedione coupled together with the same linker ()Citation220.
Scheme 48. Bisubstrate adducts for riboflavin synthase.
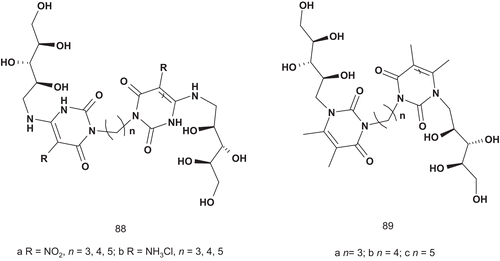
Only the bis(nitro-uracils) 88a–b and bis(luminazines) 89a–c were evaluated against riboflavin synthase and lumazine synthase.
The most potent inhibitor proved to be 89b, with a Ki value of 37 μM and a C-3 carbon chain linker. This greater relative potency suggested that 89b might act as a bisubstrate adduct inhibitorCitation220.
4.12. Nicotinamide mononucleotide adenyltransferase
Nicotinamide mononucleotide transferase (NMNAT; EC 2.7.7.1) catalyzes the synthesis of NAD+ from nicotinamide mononucleotide (NMN) and adenosine triphosphate (ATP). It is the last step in both de novo and salvage NAD+ biosynthetic pathways, and is an essential protein in all organismsCitation221. NMNAT activity has therefore been identified as a potential target for development of novel chemotherapies, and Magni et al. developed several multisubstrate adducts () as putative inhibitors of NMNATCitation222.
Scheme 49. Multisubstrates for nicotinamide mononucleotide transferase.
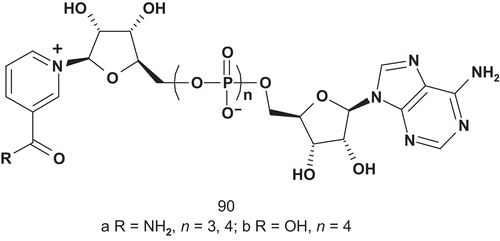
The NMNAT catalytic reaction proceeds via a ternary complex formation; therefore, the enzyme should be inhibited efficiently and specifically by multisubstrate analogs composed with NMN and AMP but connected with one more phosphoryl unit as linkerCitation223. The evaluation of these multisubstrates did not show great inhibitory activity, but has proved useful for mechanistic and metabolic studies of this enzyme in nicotinamide adenine dinucleotide (NAD) biosynthesisCitation222.
Conclusion
Over the last four decades, a number of methodologies have been developed and applied in the design of enzyme inhibitors. One such method, the MAI approach, has been used extensively and successfully in the design of inhibitors, which has allowed insights into enzymatic mechanisms and the development of drug leads. The synergistic effect of the covalent association between substrate and cofactor analogs or between multiple substrate analogs allows the creation of inhibitor templates possessing Ki values within the substrate Km range whilst also achieving satisfactory enzyme selectivity. For many enzymes for which the MAI approach has been adopted, selectivity and affinity have been greatly improved by modification of the building blocks and the linker. Furthermore, the MAI approach has permitted the synthesis of chemical entities to facilitate enzyme co- crystallization studies, providing essential structural knowledge for future inhibitor design. For instance, many research groups have successfully exploited such structural findings to identify secondary binding sites and more accurately determine the distance between substrate and cofactor binding sites to allow more specific design of linker lengths and flexibilitiesCitation224–226.
In conclusion, the MAI approach provides a reliable method for designing inhibitors, and is most effective when combined with other drug-design tools such as crystallography or modeling. This method shows itself to be extremely useful for the specific probing of biological drug targets, and thus facilitates the early steps of the associated drug discovery programs. Nevertheless, for identification of lead compounds, the MAI approach is far from being appropriate, as most compounds so far reported do not possess drug-like qualities, which are so important to successful drug discovery programs. However, the MAI approach must still be considered as a relevant tool for the development of pharmacophore building blocks, which will facilitate drug design.
Acknowledgements
European Social Funding (ESF) and DEL are gratefully thanked for the doctoral fellowship to P. B. Le Calvez.
Declaration of interest: The authors report no conflicts of interest.
References
- Broom AD. J Med Chem 1989;32:2–7.
- Wolfenden R. Accts Chem Res 1972;5:10–18.
- Heller JS, Canellakis ES, Bussolotti DL, Coward JK. Biochem Biophys Acta 1975;403:197–207.
- Shemin D. Methods Enzymol 1970;17:205–11.
- Gacond S, Frère F, Nentwich M, Fauritea JP, Frankenberg-Dinkel N, Neier N. Chem Biodivers 2007;4:189–202.
- Kang K, Fromm HJ. J Biol Chem 1995;270:15539–44.
- Heim DR, Cseke C, Gerwick BC, Murdoch MG, Green SB. Pestic Biochem Physiol 1995;53:138–45.
- Cseke C, Gerwick BC, Crouse GD, Murdoch MG, Green SB, Heim DR. Pestic Biochem Physiol 1996;55:210–17.
- Fonne-Pfister R, Chemla P, Ward E, Girardet M, Kreuz KE, Honzatko RB, et al. Proc Natl Acad Sci USA 1996;93:9431–6.
- Walters EW, Lee S, Niderman T, Bernasconi P, Subramanian MV, Siehl D. Plant Physiol 1997;114:549–55.
- Hatch MD.. Phytochemistry 1967;6:115–19.
- Hanessian S, Lu PP, Sanceau JY, Chemla P, Gohda K, Fonne-Pfister R, et al. Angew Chem Int Ed 1999;38:3160–2.
- Abu-Elheiga L, Matzuk MM, Abo-Hashema KAH, Wakil SJ. Science 2001;291:2613–16.
- Lenhard JM, Gottschalk WK. Adv Drug Delivery Rev 2002;54:1199–212.
- Levert KL, Waldrop GL, Stephens JM. J Biol Chem 2002;277:16347–50.
- Campbell JW, Cronan JE Jr. Annu Rev Microbiol 2001;55:305–32.
- Blanchard CZ, Waldrop GL. J Biol Chem 1998;273:19140–5.
- Levert KL, Grover L, Waldrop GL. Biochem Biophys Res Commun 2002;291:1213–17.
- Ravindranadh VS, Wilson DJ, Bennett EM, Boshoff HI, Celia L, Beck BJ, et al. J Med Chem 2006;49:7623–35.
- Ravindranadh VS, Boshoff HI, Qiao C, Bennett EM, Barry CE, Aldrich CC. J Med Chem 2006;49:31–6.
- Vannada J, Bennett EM, Wilson DJ, Boshoff HI, Barry CE, Aldrich CC. Org Lett 2006;8:4707–10.
- Finking R, Neumuller A, Solsbacher J, Konz D, Kretzschmar G, Schweitzer M, et al. Chem Biol Chem 2003;4:903–6.
- May JJ, Finking R, Wiegeshoff F, Weber TT, Bandur N, Koert U, et al. FEBS J 2005;272:2993–3003.
- Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE. Nat Chem Biol 2005;1:29–32.
- Miethke M, Bisseret P, Beckerin CL, Vignard D, Eustache J, Marahiel MA. FEBS J 2006;273:409–19.
- Neres JO, Labello PN, Somu RV, Boshoff HI, Wilson DJ, Vannada J, et al. J Med Chem 2008;51:5349–70.
- Wimalasena K, Wimalasena DS, Dharmasena S, Haines DC, Alliston KR. Biochemistry 1997;36:7144–53.
- Kruse LI, Kaiser C, DeWolf WE Jr, Frazee JS, Garvey E, Hilbert EL, et al. J Med Chem 1986;29:2465–72.
- Kruse LI, Kaiser C, DeWolf WE Jr, Frazee JS, Ross ST, Wawro J, et al. J Med Chem 1987;30:486–94.
- Kruse LI, DeWolf WE Jr, Chambers PA, Goodhart PJ. Biochemistry 1986;25:7271–8.
- Ross ST, Kruse LI, Ohlstein EH, Erickson RW, Ezekiel M, Flaim KE, et al. J Med Chem 1987;30:1309–13.
- Miyano K, Koshigoe T, Sakasai T, Hamano H. Chem Pharm Bull 1978;26:1465–72.
- Kruse LI, Kaiser C, Dewolf WE Jr, Finkelstein JA, Frazee JS, Hilbert EL, et al. J Med Chem 1990;33:781–9.
- Salituro FG, Demeter DA, Weintraub HJR, Lippert BJ, Resvick RJ, McDonald IA. J Med Chem 1994;37:4076–8.
- Kaustov L, Kababya S, Belakhov V, Baasov T, Shoham T, Schmidt A. J Am Chem Soc 2003;125:4662–9.
- Du S, Faiger H, Belakhov V, Baasov T. Bioorg Med Chem 1999;7:2671–82.
- Anderson L, Unger FM, eds. Bacterial Lipopolysaccharides. ACS Symposium, Series 231. Washington DC: American Chemical Society, 1983.
- Inouye M. Bacterial Outer Membrane: Biogenesis and Function. New York: Wiley, 1979.
- Ray PH. J Bacteriol 1980;141:635–44.
- Hedstrom L, Abeles R. Biochem Biophys Res Commun 1988;157:816–20.
- Sheffer-Dee-Noor S, Belakhov V, Baasov T. Bioorg Med Chem Lett 1993;3:1583–8.
- Baasov T, Sheffer-Dee-Noor S, Kohen A, Jakob A, Belakhov V. Eur J Biochem 1993;217:991–9.
- Dotson GD, Dua RK, Clemens JC, Wooten EW, Woodard RW. J Biol Chem 1995;270:13698–705.
- Korolev S, Ikeguchi Y, Skarina T, Beasley S, Arrowsmith C, Edwards A, et al. Nat Struct Biol 2001;9:27–31.
- Woster PM, Black AY, Duff KJ, Coward JK, Pegg AE. J Med Chem 1989;32:1300–7.
- Tang KC, Mariuzza R, Coward JK. J Med Chem 1981;24:1277–84.
- Orr GR, Danz DW, Pontoni G, Prabhakaran PC, Gould SJ, Coward JK. J Am Chem Soc 1988;110:5791–9.
- Jencks WP. Adv Enzymol Relat Areas Mol Biol 1975;45:219–23.
- Lakanen JR, Pegg AE, Coward JK. J Med Chem 1995;38:2714–27.
- Dev IK, Harvey RJ. J Biol Chem 1978;253:4242–4.
- Beardsley GP, Taylor EC, Shih C, Poore GA, Grindey GB, Moran RG. Proc AACR 1986;27:259 (abstr 1027).
- Inglese J, Blatchly RA, Benkovic SJ. J Med Chem 1989;32:937–40.
- Greasley SE, Yamashita M, Kai H, Benkovic SJ, Boger DL, Wilson IA. Biochemistry 1999;38:16783–93.
- Greasley SE, Marsilje TH, Kai H, Baker S, Benkovic SJ, Boger DL, et al. Biochemistry 2001;40:13538–47.
- Wall N, Shim JH, Benkovic SJ. J Med Chem 1999;42:3421–4.
- Klein C, Chen P, Arevalo JH, Stura EA, Marolewski A, Warren MS, et al. J Mol Biol 1995;249:153–75.
- Diasio RB, Schuetz JD, Banks W, Wallace H. Proc Am Assoc Cancer Res 1982;23:214.
- Cohen SS. Ann NY Acad Sci 1971;186:292–301.
- Srinivasan A, Amarnath V, Broom AD, Zou FC, Cheng YC. J Med Chem 1984;27:1710–17.
- Koyama H, Ayusawa D, Tsuji M, Seno T. Mutat Res 1982;105:433–8.
- Park JS, Chang CTC, Mertes MP. J Med Chem 1979;22:1134–7.
- Yang IY, Slusher RM, Broom AD, Ueda T, Cheng YC. J Med Chem 1988;31:2126–38.
- Lovelace LL, Gibson LM, Lebioda L. Biochemistry 2007;46:2823–30.
- Enkvist E, Lavogina D, Raidaru G, Vaasa A, Viil I, Lust M, et al. J Med Chem 2006;49:7150–9.
- Goldberg AR, Wong TW. Adv Enzyme Regul 1984;22:289–308.
- Kruse CH, Holden KG, Offen PH, Pritchard JAF, Reiman DJ, Bender PE, et al. J Med Chem 1988;31:1768–72.
- Traxler PM, Wacker O, Bach HL, Geissler JF, Kump W, Meyer T, et al. J Med Chem 1991;34:2328–37.
- Russo MW, Lukas TJ, Cohen S, Staros JV. J Biol Chem 1985;160:5205.
- Zoller MN, Nelson NC, Taylor SS. J Biol Chem 1981;256:10837–42.
- Kamps MP, Taylor SS, Sefton BM. Nature 1984;310:589–92.
- Baginski, I, Commerçon A, Tocqué B, Colson G, Zerial A. Biochem Biophys Res Commun 1989;165:1324–30.
- Imoto M, Umezawa K, Sawa T, Takeuchi T, Umezawa H. Biochem Int 1987;15:989–95.
- Lienhard GE, Secemski II. J Biol Chem 1973;248:1121–3.
- Miron S, Munier-Lehmann H, Craescu CT. Biochemistry 2004;43:67–77.
- Lavie A, Konrad M, Brundiers R, Goody RS, Schlichting I, Reinstein J. Biochemistry 1998;37:3677–86.
- Bone R, Cheng YC, Wolfenden R. J Biol Chem 1986;261:5731–5.
- Bone R, Cheng YC, Wolfenden R. J Biol Chem 1986;261:16410–13.
- Ikeda S, Chakravarty R, Ives DH. J Biol Chem 1986;261:15836–43.
- Shen W, Willis D, Zhang Y, Schlattner U, Wallimann T, Molloy GR. Biochem J 2002;367:369–80.
- Min K-L, Steghens J-P, Henry R, Doutheau A, Collombel C. Biochim Biophys Acta 1997;1342:83–9.
- Williams DM, Jakeman DL, Vyle JS, Williamson MP, Blackbum GM. Bioorg Med Chem Lett 1998;8:2603–8.
- Blackburn GM, Jakeman DL, Ivory AJ, Williamson MP. Bioorg Med Chem Lett 1994;4:2573–8.
- Jakeman DL, Ivory AJ, Williamson MP, Blackburn GM. J Med Chem 1998;41:4439–52.
- Shiota T. Chemistry and Biochemistry of Folates. New York: John Wiley & Sons, 1984:121.
- Shi G, Blaszczyk J, Ji X, Yan H. J Med Chem 2001;44:1364–71.
- Nakadate T, Jeng AY, Blumberg PM. Biochem Pharmacol 1988;8:1541–5.
- Ricouart A, Gesquiere JC, Tartar A, Sergheraert C. J Med Chem 1991;34:73–8.
- Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Biochemistry 1984;23:5036–41.
- Hines AC, Cole PA. Bioorg Med Chem Lett 2004;14:2951–4.
- Hjelmquist G, Andersson J, Edlund B, Engstroom L. Biochem Biophys Res Commun 1974;61:559–63.
- de la Houssaye BA, Masaracchia RA. Anal Biochem 1983;128:54–9.
- Parang K, Till JH, Ablooglu AJ, Kohanski RA, Hubbard SR, Cole PA.Nat Struct Biol 2001;8:37–41.
- Kelly JL, Mclean EW, Crouth RC, Averett DR, Tuttle JV. J Med Chem 1995;38:1005–14.
- Wielgus-Kutrowska B, Antosiewicz JM, Dlugosz M, Holý A, Bzowska A. Biophys Chem 2007;125:260–8.
- Parks RE Jr, Stoeckler JD, Cambor C, Savarese TM, Crabtree GW, Chu S-H. Purine nucleoside phosphorylase and 5′-methylthioadenosine phosphorylase: targets of chemotherapy. In: Sartorelli A, Lazo JS, Bertino JR, eds. Molecular Actions and Targets for Cancer Chemotherapeutic Agents. New York: Academic Press, 1981:229.
- Kazmers IS, Mitchell BS, Dadonna PE, Watring LL, Townsend LB, Kelley WN. Science 1981;214:1137–9.
- Tuttle JV, Krenitsky TA. J Biol Chem 1984;259:4065–9.
- Krenitsky TA, Tuttle JV, Miller WH, Moorman AR, Orr GF, Beauchamp L. J Biol Chem 1990;265:3066–9.
- Nakamura CE, Chu S-H, Stoeckler JD, Parks RE Jr. Nucleos Nucleot Nucl 1989;8:1039–43.
- Halazy S, Ehrhard A, Danzin C. J Am Chem Soc 1991;113:315–17.
- Ealick SE, Babu YS, Bugg CE, Erion MD, Guida WC, Montgomery JA, et al. Proc Natl Acad Sci USA 1991;88:11540–4.
- Kelley JL, Linn JA, McLean EW, Tuttle JV. J Med Chem 1993;36:3455–63.
- Mannisto PT, Ulmanen I, Lundström K, Taskinen J, Tenhunen J, Tilgmann C, et al. Prog Drug Res 1992;39:291–350.
- Nackley AG, Tan KS, Fecho K, Flood P, Diatchenko L, Maixner W. Pain 2007;128:199–208.
- Anderson GL, Bussolitti DL, Coward JK. J Med Chem1981;24:1271–7.
- Burns MR, Coward JK. Bioorg Med Chem 1996;4:1455–70.
- Borgulya J, Da Prada M, Dingemanse R, Scherschlich B, Schläppi B, Zücher G. Drugs Future 1991;16:719–21.
- Masjost B, Ballmer P, Borroni E, Zürcher G, Winkler FK, Jakob-Roetne R, et al. Chem Eur J 2000;6:971–82.
- Vidgren J, Svensson LA, Liljas A. Nature 1994;368:354–8.
- Lerner C, Masjost B, Ruf A, Gramlich V, Jakob-Roetne R, Zürcher G, et al. Org Biomol Chem 2003;1:42–9.
- Lerner C, Ruf A, Gramlich V, Masjost B, Zürcher G, Jakob-Roetne R, et al. Angew Chem 2001;40:4040–2.
- Paulini R, Lerner C, Jakob-Roetne R, Zürcher G, Borroni E, Diederich F. Chembiochem 2004;5:1270–4.
- Smith KS, Smith PL, Heady TN, Trugman JM, Harman WD, Macdonald TL. Chem Res Toxicol 2003;16:123–8.
- Korlipara LVP, Cooper JM, Schapira AHV. Neuropharmacology 2004;46:562–9.
- Bonifacio MJ, Archer M, Rodrigues ML, Matias PM, Learmonth DA, Carrondo MA, et al. Mol Pharmacol 2002;62:795–805.
- Kappler F, Hai TT, Hampton A. J Med Chem 1986;29:318–22.
- Kappler F, Vrudhula VM, Hampton A. J Med Chem 1987;30:1599–603.
- Vrudhula VM, Kappler F, Afshar C, Ginell SL, Lessinger L, Hampton A. J Med Chem 1989;32:885–90.
- Kappler F, Vrudhula VM, Hampton A. J Med Chem 1988;31:384–9.
- Kappler F, Hampton A. J Med Chem 1990;33:2545–51.
- Benghiat E, Crooks PA. J Med Chem 1983;26:1470–7.
- Cole C, Foster AJ, Freeman S, Jaffar M, Murray PE, Stratford IJ. Anticancer Drug Des 1999;14:383–92.
- Haraguchi M, Miyadera K, Uemura K, Sumizawa T, Furukawa T, Yamada K, et al. Nature 1994;368:198.
- Takeuchi M, Otsuka T, Matsui N, Asai K, Hirano T, Moriyama A, et al. Arthritis Rheum 1994;37:662–72.
- Creamer D, Jaggar R, Allen M, Bicknell R, Barker J. Br J Dermatol 1997;137:851–5.
- Iltzsch MH, el Kouni MH, Cha S. Biochemistry 1985;24:6799–807.
- Balzarini J, Degrève B, Estan-Gamboa A, Esnouf R, De Clercq E, Engelborghs Y, et al. FEBS Lett 2000;483:181–5.
- Allan AL, Gladstone PL, Price ML, Hopkins SA, Juarez JC, Donate F, et al..J Med Chem 2006;49:7807–15.
- Matsushita S, Nitanda T, Furukawa T, Sumizawa T, Tani A, Nishimoto K, et al. Cancer Res 1999;59:1911–16.
- Palcic MM, Heerze LD, Srivastava OP, Hindsgaul O. J Biol Chem 1989;264:17174–81.
- Hashimoto H, Endo T, Kajihara Y..xxx.J Org Chem 1997;62:1914–15.
- Gautier-Lefebvre I, Behr JB, Guillerma G, Ryder NS. Bioorg Med Chem Lett 2000;10:1483–6.
- Lau JTY, Wuensch SA. In: Ernst B, Hart GW, Sinaÿ P, eds. Carbohydrates in Chemistry and Biology. Weinheim: Wiley-VCH, 2000;3:213–26.
- Stolz F, Blume A, Hinderlich S, Reutter W, Schmidt RR. Eur J Org Chem 2004;15:3304–12.
- Schafer A, Thiem J. J Org Chem 2000;65:24–9.
- Luengo JI, Gleason JG. Tetrahedron Lett 1992;33:6911–14.
- Orbe M, Claesson A. Eur J Med Chem 1989;24:447–51.
- Norbeck DW, Kramer JB, Lartey PA. J Org Chem 1987;52:2174–9.
- Schmidt RR, Frische K. Bioorg Med Chem Lett 1993;3:1747–50.
- Vaghefi MM, Bernacki RJ, Dalley NK, Wilson BE, Robins RK. J Med.Chem 1987;30:1383–91.
- Schworer R, Schmidt RR. J Am Chem Soc 2002;124:1632–7.
- Hinou H, Sun X-L, Ito Y. J Org Chem 2003;68:5602–13.
- Izumi M, Wada K, Yuasa H, Hashimoto H. J Org Chem 2005;70:8817–24.
- Izumi M, Kaneko S, Yuasa H, Hashimoto H. Org Biomol Chem 2006;4:681–90.
- Brockhausen I, Carver JP, Schachter H. Biochem Cell Biol 1988;66:1134–51.
- Cummings RD, Trowbridge IS, Kornfeld S. J Biol Chem 1982;257:13421–7.
- Dennis JW, Granovsky M, Warren CE. Biochim Biophys Acta 1999;1473:21–34.
- Dennis JW, Laferte S, Waghorne C, Breitman ML, Kerbel RS. Science 1987;236:582–5.
- Ihara S, Miyoshi E, Ko J, Murata K, Nakahara S, Honke K, et al. J Biol Chem 2002;277:16960–7.
- Demetriou M, Nabi IR, Oppolino M, Dedhar S, Dennis JW. J Cell Biol 1995;130:383–92.
- Guo HB, Lee I, Kamar M, Akiyama SK, Pierce M. Cancer Res 2002;62:6837–45.
- Saito T, Miyoshi E, Sasai K, Nakano N, Eguchi H, Honke K, et al. J BiolChem 2002;277:17002–8.
- Hanashima S, Manabe S, Inamori KI, Taniguchi N, Ito Y. Chem Eur J 2006;12:3449–62.
- Tvaroska I, André I, Carver JP. J Am Chem Soc 2000;122:8762–76.
- Tahir SH, Hindsgaul O. Can J Chem 1986;64:1771–80.
- De Angelis J, Gastel J, Klein DC, Cole PA. J Biol Chem 1998;273:3045–50.
- Kim CM, Cole PA. J Med Chem 2001;44:2479–85.
- Khalil EM, Cole PA. J Am Chem Soc 1998;120:6195–6.
- Khalil EM, De Angelis J, Ishii M, Cole PA. Proc Natl Acad Sci USA 1999;96:12418–23.
- Zheng W, Cole PA. Bioorg Chem 2003;31:398–411.
- Frisch SM, Mymryk JS. Nat Rev Mol Cell Biol 2002;3:441–52.
- Timmermann S, Lehrmann H, Polesskaya A, Harel-Bellan A. Cell Mol Life Sci 2001;58:728–36.
- Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin SF, et al. Nat Genet 2000;24:300–3.
- Deguchi K, Ayton PM, Carapeti M, Kutok JL, Snyder CS, Williams IR, et al. Cancer Cell 2003;3:259–71.
- Lau OD, Kundu TK, Soccio RE, Ait-Si-Ali S, Khalil EM, Vassilev A, et al. Mol Cell 2000;5:589–95.
- Lau OD, Courtney AD, Vassilev A, Marzilli LA, Cotter RJ, Nakatani Y, et al. J Biol Chem 2000;275:21953–9.
- Thompson PR, Kurooka H, Nakatani Y, Cole PA. J Biol Chem 2001; 276:33721–9.
- Sagar V, Zheng W, Thompson PR, Cole PA. Bioorg Med Chem 2004;12:3383–90.
- Cebrat M, Kim CM, Thompson PR, Daugherty M, Cole PA. Bioorg Med Chem 2003;11:3307–13.
- Wright GD, Berghuis AM, Mobashery S. Aminoglycoside antibiotics: structures, functions and resistance. In: Rosen BP, Mobashery S, eds. Resolving the Antibiotic Paradox: Progress in Understanding Drug Resistance and Development of New Antibiotics. New York: Plenum, 1998:27.
- Magnet S, Blanchard JS. Chem Rev 2005;105:477–97.
- Auclair K, Gao F, Yan X. US Patent Application 11/359 274, filed February 21 2006.
- Gao F, Yan X, Baettig OM, Berghuis AM, Auclair K. Angew Chem Int Ed 2005;44:6859–62.
- Wright GD, Ladak P. Antimicrob Agents Chemother 1997;41:956–60.
- Gao F, Yan X, Shakya T, Baettig OM, Ait-Mohand-Brunet S, Berghuis AM, et al. J Med Chem 2006;49:5273–81.
- Liu M, Haddad J, Azucena E, Kotra LP, Kirzhner M, Mobashery S. J Org Chem 2000;65:7422–31.
- Lipscomb WN. Adv Enzymol Relat Areas Mol Biol 1994;68:67–151.
- Laing N, Chan WW-C, Hutchinson DW, öberg B. FEBS Lett 1990;260:206–8.
- Dutta PL, Foye WO. J Pharm Sci 1990;79:447–52.
- Swyryd EA, Seaver SS, Starkv GR. J Biol Chem 1974;246:6945–50.
- Collins KD, Startk GR. J Biol Chem1971;246:6599–605.
- Newell JO, Markby DW, Schachman HK. J Biol Chem 1989;264:2476–81.
- Howlett GJ, Schachman HK. Biochemistry 1977;16:5077–83.
- Endrizzi JA, Beernink PT, Alber T, Schachman HK. Proc Natl Acad Sci USA 2000;97:5077–82.
- Van Boxstael S, Cunin R, Khan S, Maes D. J Mol Biol 2003;326:203–16.
- Krause KL, Volz KW, Lipscomb WN. Proc Natl Acad Sci USA 1985;82:1643–7.
- Grison C, Coutrot P, Comoy C, Balas L, Joliez S, Lavecchia G, et al. Eur J Med Chem 2004;39:333–44.
- Bowman KG, Bertozzi CR. Chem Biol 1999;6:R9–22.
- Kehoe JW, Bertozzi CR. Chem Biol 2000;7:R57–61.
- Falany CN. FASEB J 1997;11:206–16.
- Kakuta Y, Pedersen LG, Pedersen LC, Negishi M. Trends Biochem Sci 1998;23:129–30.
- Armstrong JI, Ge X, Verdugo DE, Winans KA, Leary JA, Bertozzi CR. Org Lett 2001;3:2657–60.
- Armstrong JI, Verdugo DE, Bertozzi CR. J Org Chem 2003;68:170–3.
- Zhang FL, Casey PJ. Annu Rev Biochem 1996;65:241–69.
- Fu HW, Casey PJ. Recent Prog Horm Res 1999;54:315–43.
- Reiss Y, Goldstein JL, Seabra MC, Casey PJ, Brown MS. Cell 1990;62:81–8.
- Gibbs JB. Cell 1991;65:1–4.
- Graham SL, deSolms SJ, Guiliani EA, Kihl NE, Mosser SD, Oliff AI, et al. J Med Chem 1994;37:725–32.
- Pompliano DL, Rands E, Schaber MD, Mosser SD, Anthony NJ, Gibbs JB. Biochemistry 1992;31:3800–7.
- Singh SB, Zink DL, Liesch JM, Goetz MA, Jenkins RG, Nallin-Olmstead M, et al. Tetrahedron 1993;49:5917–26.
- Bhide RS, Patel DV, Patel MM, Robinson SP, Hunihan LW, Gordon EM. Bioorg Med 2Chem Lett 1994;4:2107–12.
- Patel DV, Gordon EM, Schmidt Harold RJ, Weller N, Young MG, Zahler R, et al. J Med Chem 1995;38:435–42.
- Patel DV, Young MG, Robinson SP, Hunihan LW, Dean BJ, Gordon EM. J Med Chem 1996;39:4197–210.
- Reynolds JEF, ed. Martindale: The Extra Pharmacopoeia, 31st ed. London: Royal Pharmaceutical Society of Great Britain, 1996:821.
- Schlitzer M, Sattler I. Eur J Med Chem 2000;35:721–6.
- Schlitzer M, Böhm M, Sattler I, Dahse HM. Bioorg Med Chem 2000;8:1991–2006.
- Schlitzer M, Böhm M, Sattler I. Bioorg Med Chem 2000;8:2399–406.
- Schlitzer M, Sattler I. Bioorg Med Chem 1999;7:2391–5.
- Mitsch A, Bergemann S, Gust R, Sattler I, Schlitzer M. Arch Pharm Pharm Med Chem 2003;336:242–50.
- Schlitzer M, Böhm M, Sattler I. Bioorg Med Chem 2002;10:615–20.
- Zhang H. Structure 2003;11:237–9.
- Reid TS, Terry KL, Casey PJ, Beese LS. J Mol Biol 2004;343:417–33.
- Casey P, Thissen JA, Moomaw JF. Proc Natl Acad Sci USA 1991;88:8631–5.
- Yokoyama K, Goodwin GW, Ghomaschi F, Glomset JA, Gelb MH. Proc Natl Acad Sci USA 1991;88:5302–6.
- El Oualid F, Baktawar J, Leroy IM, van den Elst H, Cohen LH, van der Marel GA, et al. Bioorg Med Chem 2005;13:1463–75.
- El Oualid F, Bruining L, Leroy IM, Cohen LH, van Boom JH, van der Marel GA, et al. Helv Chim Acta 2002;85:3455–72.
- Plaut GWE, Smith CM, Alworth WL. Annu Rev Biochem 1974;43:899–922.
- Plaut GWE. In: Florkin M, Stotz EH, eds. Comprehensive Biochemistry. Amsterdam: Elsevier, 1971;21:11.
- Bacher A, Ladenstein R. In: Müller F, ed. Chemistry and Biochemistry of Flavoenzymes. Boca Raton, FL: Chemical Rubber Co., 1991;II:293.
- Cushman M, Mavandadi F, Yang D, Kugelbrey K, Kis K, Bacher A. J Org Chem 1999;64:4635–42.
- Saridakis V, Christendat D, Kimber MS, Dharamsi A, Edwards AM, Pai EF. J Biol Chem 2001;276:7225–32.
- Sorci L, Cimadamore F, Scotti S, Petrelli R, Cappellacci L, Franchetti P, et al. Biochemistry 2007;46:4912–22.
- Cleland WW. Adv Enzymol Relat Areas Mol Biol 1977;45:273–87.
- Bose R, Holbert MA, Pickin KA, Cole PA. Curr Opin Struct Biol 2006;16:668–75.
- Vazquez J, Tautz L, Ryan JJ, Vuori K, Mustelin T, Pellecchia M. J Med Chem 2007;50:2137–43.
- Hines AC, Parang K, Kohanski RA, Hubbard SR, Cole PA. Bioorg Chem 2005;33:285–97.