Abstract
Barlerisides A (1) and B (2), new phenolic glycosides, have been isolated from the n-butanol soluble sub-fraction of Barleria acanthoides along with two known compounds acteoside (3) and p-hydroxycinnamic acid (4). Their structures have been assigned on the basis of spectral studies. Both 1 and 2 showed significant activity in the superoxide scavenging assay while weak inhibitory activity was observed against the enzyme xanthine oxidase.
Introduction
The genus Barleria belongs to the Acanthaceae family. It comprises 230 species, occurring mainly in tropical and subtropical regions. It is represented in Pakistan by four speciesCitation1. Barleria acanthoides Vahl occurs in tropical Africa and AsiaCitation2. In Pakistan it is very common in the outskirts of Karachi cityCitation3. The plant is well known for its anti-inflammatory properties. The seeds are used as an antidote for snake-bite, and the roots and leaves are used to reduce swellings and an infusion is given in cough. It is reported for its use in diarrhea and also as a diaphoretic and an expectorantCitation2. No phytochemical studies have so far been carried out on this species. The methanolic extract of Barleria acanthoides showed strong toxicity in a brine shrimp lethality testCitation4. On further fractionation, the major toxicity was observed in the n-butanolic sub-fraction. This prompted us to carry out studies on this fraction, resulting in the isolation and structural elucidation of two new phenolic glycosides named as barlerisides A (1) and B (2), along with two known compounds acteoside (3)Citation5 and p-hydroxycinnamic acid (4)Citation6. Both compounds 1 and 2 showed significant activity in a superoxide scavenging assay, while they also showed weak inhibitory activity against the enzyme xanthine oxidase.
Material and methods
Plant material
The whole plant of Barleria acanthoides Vahl was collected in 2007 from Karachi, Pakistan and identified by Dr. Suraiya Khatoon, Plant Taxonomist, Department of Botany, University of Karachi, Pakistan, where a voucher specimen (KUH-3969) has been deposited.
General experimental procedures
Optical rotations were measured on an Atago AP-300 digital polarimeter using a 200 mm tube. Ultraviolet (UV) and infrared (IR) spectra were recorded on Hitachi-UV-3200 and Jasco-320-A spectrometers. Electron ionization (EIMS), fast atom bombardment (FAB-MS), and high-resolution FAB (HR-FAB-MS) spectra (neg. mode; matrix: glycerol) were recorded on Jeol JMS-HX 110 and JMS-DA 5000 mass spectrometers. 1H and 13C nuclear magnetic resonance (NMR), heteronuclear multiple quantum coherence (HMQC), and heteronuclear multiple bond coherence (HMBC) spectra were recorded on a Bruker spectrometer operating at 400 MHz for 1H- and 100 MHz for 13C-NMR, respectively. The chemical shifts are reported in ppm (δ), and the coupling constants (J) are reported in Hz. Column chromatography was carried out on various adsorbents including Diaion HP-20 ion exchange resin (Nippon Rensui Co., Japan) and polyamide 6 powder (0.05–0.16 mesh; Serva, Belgium). Thin layer chromatography (TLC) was performed on pre-coated silica gel 60 F254 plates (20 × 20 cm, 0.2 mm thick; E. Merck, Darmstadt, Germany), and visualization was achieved at 254 nm and also by spraying with ceric sulfate reagent. Gas chromatography (GC) was run on a Shimadzu GC-4CM instrument with a flame ionization detector. High performance liquid chromatography (HPLC) was used for final purification on a recycling preparative system (LC-908W-C-60; Japan Analytical Industry Co. Ltd) using a column of ODS-M-80 (4 μm (250 × 20 mm)).
Extraction and isolation
Shade-dried whole plant material (20 kg) of Barleria acanthoides was extracted at room temperature with methanol (3 × 50 mL). The combined methanolic extract (300 g) was divided into n-hexane (100 g), EtOAc (60 g), n-BuOH (23 g), and water soluble (95 g) fractions. The n-butanolic fraction was dissolved in water and column-chromatographed over Diaion HP-20, eluting successively with H2O, H2O–MeOH (1:1), and MeOH. The fraction that eluted with MeOH–H2O (1:1) was further chromatographed over polyamide, eluting with CH2Cl2–MeOH to obtain major sub-fractions I–III. Fraction I obtained from CH2Cl2–MeOH (9.8:0.2) was subjected to HPLC with flow rate 3 mL/min, using MeOH–H2O (1:1) as eluant, to afford 4 (15.5 mg, tR 21 min). Fraction II obtained from CH2Cl2–MeOH (9.6:0.4) was also subjected to HPLC with flow rate 3.5 mL/min, using MeOH–H2O (2:1) as eluant, to obtain 1 (12 mg, tR 28 min). Fraction III obtained from CH2Cl2–MeOH (9:1) was further fractionated by HPLC into two fractions, IIIA and IIIB. Fraction IIIA was finally purified by HPLC using MeOH–H2O (1:1) as eluant, to furnish 2 (10 mg, tR 32 min). The purification of fraction IIIB by HPLC using MeOH–H2O (1:1) as eluant yielded 3 (4 mg, tR 38 min).
Compounds 3 and 4 were respectively identified as acteoside and p-hydroxycinnamic acid through comparison of their physical and spectral data with those reported in the literatureCitation5,Citation6.
Compound 1
Gummy solid; (12 mg). [α]D25 −49.0 (c 0.001, MeOH); UV (MeOH) λmax (log ϵ): 270 (4.41) and 336 (4.48) nm; IR (KBr) λmax: 3400, 1685, 1620, and 1172 cm−1; EIMS m/z 270 [M–sugar]+, 152 [C7O4H4]+, 118 [C8OH6]+; HR-FAB-MS (neg.): m/z 431.0941 [M–H]−, (calcd. for C21H19O10: 431.0978); 1H-NMR (400 MHz C5D5N) δ: 4.19 (1H, dd, J = 11.3, 2.5 Hz, H-6b″), 4.20 (1H, m, H-5″), 4.35 (1H, m, H-2″), 4.37 (1H, m, H-4″), 4.40 (1H, m, H-3″), 4.57 (1H, dd, J = 11.3, 4.9 Hz, H-6″a), 5.81 (1H, d, J = 7.2 Hz, H-1″), 6.83 (1H, d, J = 1.8 Hz, H-5), 6.87 (1H, s, H-3), 7.08 (1H, d, J = 1.8 Hz, H-7), 7.22 (2H, d, J = 9.0 Hz, H-3′/5′), 7.86 (2H, d, J = 9.0 Hz, H-2′/6′).13C-NMR (100 MHz C5D5N) δ: 162.6 (C-2), 104.0 (C-3), 182.8 (C-4), 100.6 (C-5), 164.0 (C-6), 95.3 (C-7), 162.9 (C-8), 157.9 (C-9), 106.5 (C-10), 122.0 (C-1′), 128.9 (C-2′/6′), 116.9 (C-3′/5′), 164.9 (C-4′), 101.8 (C-1″), 74.8 (C-2″), 78.5 (C-3″), 71.1 (C-4″), 79.2 (C-5″), 62.3 (C-6″).
Compound 2
Gummy solid; (10 mg). [α]D25 −45 7 (c 0.001, MeOH); UV (MeOH) λmax (log ϵ): 223 (4.27), 245 (4.10), 290 (4.20), and 332 (4.25) nm. IR (KBr) λmax: 3400, 1694, 1630, 1606, and 1518 cm−1. HR-FAB-MS (neg.): m/z 623.1940 [M–H]−, (calcd. for C29H35O15: 623.1976). 1H-NMR (400 MHz CD3OD) δ: 1.24 (3H, d, J = 6.1, H-6″′), 2.77 (2H, t, J = 8.5 Hz, H-β), 3.39 (1H, m, H-4), 3.41 (1H, m, H-5″′), 3.44 (1H, m, H-4″′), 3.53 (1H, m, H-2), 3.55 (1H, m, H-5), 3.74 (1H, m, H-2″′), 3.76 (1H, m, H-3), 3.90 (2H, m, H-α), 3.93 (1H, m, H-3″′), 4.31–4.36 (2H, m, H-6), 4.47 (1H, d, J = 8.0 Hz, H-1), 5.16 (1H, d, J = 1.0 Hz, H-1″′), 6.26 (1H, d, J = 15.8 Hz, H-7″), 6.51 (1H, dd, J = 8.0,1.7 Hz, H-6′), 6.63 (1H, d, J = 1.7 Hz, H-2′), 6.66 (1H, d, J = 8, H-5′), 6.67 (1H, d, J = 8.0 Hz, H-5″), 6.89 (1H, dd, J = 8.0, 1.14 Hz, H-6″), 7.02 (1H, d, J = 1.8 Hz, H-2″), 7.54 (1H, d, J = 15.8 Hz, H-8″). 13C-NMR (100 MHz CD3OD) δ: 131.3 (C-1′),117.0 (C-2′),144.4 (C-3′), 146.8 (C-4′), 116.5 (C-5′), 121.2 (C-6′), 72.2 (C-α), 36.7 (C-β), 127.5 (C-1″), 114.9 (C-2″), 148.8 (C-3″), 146.1 (C-4″), 116.3 (C-5″), 123.1 (C-6″), 114.7 (C-7″), 147.3 (C-8″), 169.1 (C-9″), 104.4 (C-1), 75.4 (C-2), 83.8 (C-3), 70.3 (C-4), 75.7 (C-5), 64.6 (C-6), 102.7 (C-1″′), 72.3 (C-2″′), 72.2 (C-3″′), 73.0 (C-4″′), 70.0 (C-5), 17.1 (C-6″′).
Acid hydrolysis of compounds 1 and 2
Compound 1 (4 mg) in MeOH (5 mL) containing 1N HCl (4 mL) was refluxed for 4 h, concentrated under reduced pressure, and diluted with H2O (8 mL). It was extracted with ethyl acetate, and the aglycone obtained through preparative TLC crystallized from CHCl3 as yellow needles, m.p. 300°C. Its color reactions, melting point, and spectral data showed complete agreement with those reported in the literature for 6,8,4′-trihydroxyflavoneCitation7.
The aqueous phase was concentrated, and d-glucose was identified by the sign of its optical rotation ([α]27D = +52.2°) and further confirmed by Co-TLC with an authentic sample.
A solution of 2 (4 mg) in 10% H2SO4 (2 mL) was heated in a boiling water bath for 30 min. The solution was passed through an Amberlite IR-45 column and concentrated to give a residue, which was reduced with sodium borohydride (4 mg) for 1 h. The reaction mixture was passed through an Amberlite IR-120 column and concentrated to dryness. Boric acid was removed by distillation with MeOH and the residue was acetylated with acetic anhydride (two drops) and pyridine (two drops) at 100°C for 1 h. The reagents were evaporated in vacuo, and glucitol acetate and rhamnitol acetate were detected in a ratio of 1:1 by gas chromatography. (Conditions: column, 1.5% OV-17, 3 mm × 1.5 m; column temp., 180°C; carrier gas, N2 (30 mL/min); tR (min) 2.0 (rhamnitol acetate), 5.5 (glucitol acetate)).
Methanolysis of compound 2
Compound 2 (1 mg) was refluxed with methanolic CH3COCl (2 mL) for 30 min, and then the reagents were evaporated off in vacuo. The presence of methyl caffeate and 3′,4′-dihydroxyphenethyl alcohol in the residue was demonstrated by Co-TLC with authentic samples, using CHCl3–MeOH (20:1) as eluant (3′,4′-dihydroxyphenethyl alcohol: retardation factor (Rf) 0.06; methyl caffeate: Rf 0.20).
Superoxide scavenging assay
Superoxide scavenging activityCitation8 was assayed in phosphate buffer (0.1 M, pH 7.5). Xanthine oxidase (0.003 units/well) and the test sample in dimethylsulfoxide (DMSO) were mixed in a 96-well microtiter plate and preincubated for 10 min at room temperature, then WST-1 reagent (15 μM) was added. The reaction was initiated by adding 0.1 mM of xanthine, and uric acid was measured spectroscopically at 295 nm and the reduction of WST-1 was read at 450 nm using a Molecular Devices system.
Xanthine oxidase activity
Xanthine oxidase activity was measured by a known methodCitation8. The reaction mixture containing 10 μL of 1 mM pure sample was dissolved in DMSO, 125 μL of phosphate buffer (0.1 M, pH 7.4), 0.003 units of xanthine oxidase dissolved in buffer (25 μL), and 25 μL of 0.15 mM xanthine as substrate for the enzyme. After the addition of xanthine oxidase, the mixture was incubated for 10 min and pre-read in the UV region (λmax 295 nm). The substrate was added to the reaction mixture, and continuous reading for 30 min at 1 min intervals was carried out. The percentage inhibitory activity by the samples was determined against a DMSO blank and calculated using the following formula:
% Inhibition = 100 − [(OD test compound/OD control) × 100]
where OD is optical density. The IC50 of samples was determined using EZ-Fit Windows-based software.
Results and discussion
The methanolic extract of Barleria acanthoides was divided into n-hexane, ethyl acetate, n-butanol, and water soluble sub-fractions. The n-butanolic sub-fraction was subjected to a series of chromatographic techniques, including recycling HPLC, to obtain compounds 1–4, and their structures were established by UV, IR, mass (MS), and NMR spectroscopy.
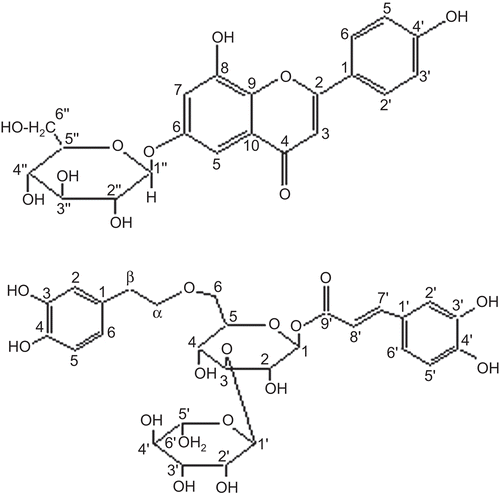
Barleriside A (1) () was isolated as a yellow gummy solid, and gave violet coloration with FeCl3 and a red color in the Shinoda test, typical for flavonoidsCitation9. The molecular formula was assigned as C21H20O10 by HR-FAB-MS in negative mode, showing the [M–H]− peak at m/z 431.0941 (calcd. for C21H19O10, 431.0978). It further showed a prominent fragment at m/z 270 due to loss of the hexose unit. EI-MS gave the peak of the aglycone at m/z 270, followed by retro-Diels–Alder (RDA) fragments at m/z 152 and 118, confirming the presence of two hydroxyl groups in ring A along with one hydroxyl group in ring B. The IR spectrum showed the presence of hydroxyl groups (3400 cm−1), conjugated carbonyl (1685 cm−1), and a double bond (1620 cm−1). The UV spectrum showed absorption maxima at 270 and 336 nm, characteristic of a flavonoidCitation10. The 1H-NMR spectrum provided meta coupled protons of ring A at δ 6.83 (1H, d, J = 1.8 Hz, H-5) and 7.08 (1H, d, J = 1.8 Hz, H-7). The B ring gave a typical AA′ BB′ pattern at δ 7.86 (2H, d, J = 9.0 Hz, H-2′/6′) and 7.22 (2H, d, J = 9.0 Hz, H-3′/5′), confirming the presence of a hydroxyl group at C-4′. The signal of an anomeric proton was observed at δ 5.81 (1H, d, J = 7.2 Hz, H-1″). Its larger coupling constant allowed us to assign β-linkage to the sugar moiety. The other oxymethine protons of the hexose moiety were observed between δ 4.20 and 4.40 and oxymethylene protons were observed at δ 4.57 (1H, dd, J = 11.3, 4.9 Hz, H-6″a) and 4.19 (1H, dd, J = 11.3, 2.5 Hz, H-6″b).
Figure 1. Structures of compounds 1 and 2.
The 13C-NMR spectra (BB and DEPT) showed 21 signals comprising one methylene, 12 methine, and eight quaternary carbons. The signals at δ 162.6, 104.0, and 182.8 were typical of the flavone skeleton. Apart from other peaks of aromatic carbons, the spectrum showed signals of anomeric carbon at δ 101.8, oxymethine carbons of the sugar moiety ranging between δ 71.1 and 79.2, and oxymethylene carbon at δ 62.3.
Acid hydrolysis of (1) provided an aglycone, identified as 6,8,4′-trihydroxyflavone, which has not so far been reported from a natural source but has been previously synthesizedCitation7. The sugar could be isolated and identified as D-glucose through the sign of optical rotation, and further confirmed by Co-TLC using an authentic sample.
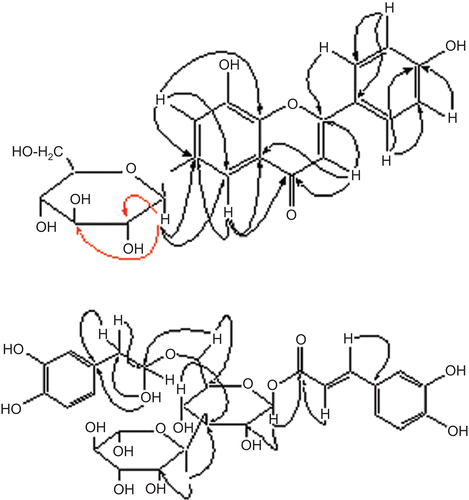
The remaining problem was to assign the point of attachment of the sugar moiety. It could be confirmed at C-6, as strong HMBC correlations () were observed between the anomeric proton at δ 5.81 and those of C-2 (δ 74.8), C-3″ (δ 78.5), and C-6 (δ 164.0), respectively. Thus, the structure of barleriside A (1) could be assigned as 8,4′-dihydroxy-6-O-(β-D-glucopyranosyl)flavone.
Figure 2. Important heteronuclear multiple bond coherence (HMBC) correlations of 1 and 2.
Barleriside B (2) () was obtained as a gummy solid and gave a brown coloration with FeCl3. The IR spectrum showed absorptions for hydroxyl groups (3400 cm−1), conjugated ester (1694 cm−1), a double bond (1630 cm−1), and aromatic rings (1606– 1518 cm−1). The UV spectrum showed absorption maxima at 223, 245sh, 290, and 332 nm. The molecular formula was established by HR-FAB-MS, showing a [M–H]− peak at m/z 623.1940 (calcd. for C29H35O15, 623.1976). The 1H-NMR spectrum showed signals due to a methyl group of rhamnose at δ 1.24 (3H, d, J = 6.1 Hz, H-6″′), two benzylic methylene protons at δ 2.77 (2H, t, J = 8.5 Hz, H-β) and 3.90 (2H, m, H-α), one glucose anomeric proton at δ 4.47 (1H, d, J = 8.0 Hz, H-1), a rhamnose anomeric proton at δ 5.16 (1H, d, J = 1.0 Hz, H-1″′), trans-olefinic protons at δ 6.26 (1H, d, J = 15.8 Hz, H-7″) and 7.54 (1H, d, J = 15.8 Hz, H-8″), and aromatic protons at δ 6.63–7.02 (6H).
The broadband and DEPT 13C-NMR spectra showed 29 signals comprising one methyl, three methylene, 18 methine, and seven quaternary carbons. The carbonyl carbon resonated at δ 169.1 while olefinic carbons were observed at δ 114.7 and 147.3, respectively. Two oxymethylene carbons were observed at δ 64.6 and 72.2, while oxymethine carbons of the sugar unit were observed between δ 70.0 and 83.8. Four oxygenated aromatic carbons appeared at δ 148.8, 146.8, 146.1, and 144.4.
Acid hydrolysis of 2 with 10% H2SO4 afforded D-glucose and l-rhamnose in a ratio of 1:1.On the other hand, methanolysis of 2 with acetylchloride in methanol afforded methyl caffeate and 3′,4′-dihydroxyphenethyl alcohol. The above data for 2 closely resemble those of acetosideCitation5. However, the two compounds differ widely in value of specific rotation; therefore, the former must be a positional isomer of acetoside. The individual signals of the glucose unit were carefully identified through 1H–1H correlation spectroscopy (COSY) and HMBC correlations. Detailed HMBC experiments showed the presence of caffeic acid and phenethyl moieties at C-1 and C-6, respectively (). Compound 2 was therefore assigned the structure 1-O-caffeoyl-6-(3′,4′-dihydroxyphenyl)ethyl-O-α-L-rhamnopyranosyl-(1→3) β-d-glucopyranoside.
Inflammation occurs as a defensive response, which induces physiological adaptation to limit tissue damage and remove pathogenic infectionsCitation11. Reactive oxygen species (ROS) are formed subsequent to the assembly and activation of the phagocyte-specific enzyme, NADPH (reduced nicotinamide adenine dinucleotide phosphate) oxidase. This process is initiated by the production of superoxide anion (O2–), during a ‘respiratory burst’ of non-mitochondrial oxygen uptake by the NADPH oxidase systemCitation12.
A superoxide scavenging assay was carried out on compounds 1 and 2. Both showed significant superoxide scavenging activity. However, barleriside B (2) was more potent due to the presence of an increased number of phenolic groups as well as the caffeoyl moiety (). Caffeic acid itself is commonly cited in the literature as a potent antioxidant. On the other hand, both compounds showed comparable but weak inhibitory activity against the enzyme xanthine oxidase. The slightly lower activity of 2 might be due to the presence of an additional sugar unit.
Table 1. IC50 (μM) values of compounds 1 and 2 in the superoxide scavenging assay and also against xanthine oxidase.
Declaration of interest: The authors report no conflicts of interest.
References
- Ali SI, Nasir E. Flora of Pakistan. Karachi: Department of Botany, University of Karachi, 1989;(191):94–101.
- George W. A Dictionary of the Economic Products of India. Delhi: Periodical Experts, 1972;1:399–401.
- Jaffery SMH. Flora of Karachi. Karachi: The Book Corporation of Pakistan, 1966:314–15.
- Meyer BN, Ferrigni NR, Putnum JE, Jacobson LB, Nicholas DE, McLaughlin JL. Brine shrimp: a convenient general bioassay for active plant constituents. Planta Med 1982;45:31–4.
- Hiromi K, Hiroko K, Toshio M, Seigo F. Studies on constituents of Cistanchis Herba. Chem Pharm Bull 1984;32:3009–14.
- Sousa JPB, Filho AAS, Bueno PCP, Gregorio LE, Furtado NAJC, Jorge RF, et al. A validated reverse phase HPLC analytical method for the quantification of phenolic compounds in Baccharis dracunculifolia. Phytochem Anal 2009;20:24–32.
- Simpson TH. Derivatives of 6,8-dihydroxyflavone. J Org Chem 1963;28:2107–10.
- Ferda C.Effect of Rhus coriarial L. (Anacardiaceae) on superoxide radical scavenging and xanthine oxidase activity. J Enzyme Inhib Med Chem 2003;18:59–62.
- Sachdev K, Kulshreshta DK. Aliarin: a new flavonoid from Donaea viscose Linn. Indian J Chem B 1982; 21,798–9.
- Voirin B. UV spectral differentiation of 5-hydroxy and 5-hydroxy-3-methoxyflavones with mono- (4’), di- (3’,4’) or tri (3’,4’,5’)-substituted B rings. Phytochemistry 1983;22:2107–45.
- Russin A, Cabec VL, Lonchampt M, De Naday J, Canet E, Parini IM. Neutrophil-associated inflammatory responses in rats are inhibited by phenylarsine oxide. Eur J Pharmacol 1997;322:91–6.
- Tan AS, Berridge VM. Superoxide produced by activated neutrophils efficiently reduces the tetrazolium salt, WST-1 to produce a soluble formazan: a simple calorimetric assay for measuring respiratory burst activation and for screening anti-inflammatory agents. J Immunol Methods 2000;238:50–68.