
Abstract
Among its various catalytic activities, “chymotrypsin-like” activity of proteasome, a large multicatalytic proteinase complex has emerged as the focus of drug discovery efforts in cancer therapy. Herein, we report results from our investigation on a series of peptidomimetic inhibitors.
Proteasome is a large multicatalytic proteinase complex that plays an important role in several critical cellular functions, including the processing of proteins involved in cell cycle progression and gene expressionCitation1–5. The 26S form of the proteasome, responsible for the ATP-dependent degradation of poly-ubiquitinated proteins, is composed of one copy of the catalytic core, known as the 20S subunit and two copies of the 19S regulatory subunit. The X-ray crystal structure of the 20S subunit from an archaebacterial proteasome revealed it to be a barrel-shaped structure made up of four stacked ringsCitation6. The two outer rings are each composed of seven identical α-subunits and the two inner rings are each composed of seven identical β-subunits. The catalytic nucleophile is the hydroxyl group of the N-terminal threonine (Thr) of the β-subunit; thus, this enzyme is the first reported member of the threonine proteinase class. The proteasome from higher organisms has the same quaternary structureCitation7; however, the seven α- and seven β-subunits are distinct from one another. The 20S proteasome has been shown to possess multiple catalytic activities, including chymotryptic, tryptic and peptidylglutamyl-like activities; thus, the complex is capable of cleaving most peptide bonds. However, among its various activities, the “chymotrypsin-like” activity of the proteasome has emerged as the biological function of greatest interest and the focus of drug discovery efforts. Increased levels of this enzyme and subsequent protein breakdown have been implicated in many disease states, including muscular dystrophy, cachexia accompanying cancer and malnutrition, lupus and cancers such as acute leukemiaCitation8–11. Proteasome also controls levels of proteins critical for cell cycle control, including p53, p27 and cyclin B, and is responsible for activation of the transcription factor NF-κB through the degradation of its regulatory subunit IκBαCitation12. Thus, development of proteasome inhibitors has emerged as an attractive target for cancer therapyCitation13–15. Previously, relying on a substrate-based approach, our laboratories reported on novel, potent and selective peptidomimetic inhibitors of the chymotrypsin (CT)-like activity of proteasome containing an aldehyde enzyme reactive group at the P1-position (, compounds 1 and 2, IC50 values of 2 and 6 nM, respectively)Citation16,Citation17. The team subsequently investigated additional potential enzyme reactive groups, structural variants spanning P3–P4 region as well as the nature of the protective group on P2-arginine moiety in this class of peptidomimetic inhibitors.
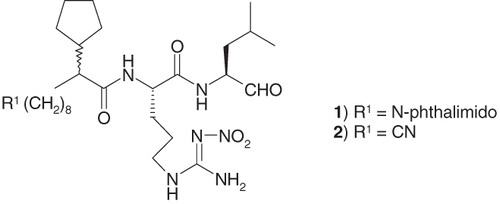
Figure 1. Structures of compounds 1 and 2.
Coupling of compound 2 () with methoxylamine hydrochloride in the presence of pyridine generated compound 3 () in 70% yield (). Similar coupling of compound 2 with a carboxamide-substituted hydrazine derivative in the presence of sodium acetate produced compound 4 () in 60% yield ().
Scheme 1. Reagents and conditions: (a) for compound 3: compound 2, MeO–NH2·HCl, pyridine, room temperature to 80 °C, 1 h, 70%; (b) for compound 4: compound 2, H2NC(=O)–NH–NH2·HCl, Na-acetate, ethanol–water, room temperature, overnight, 60%.
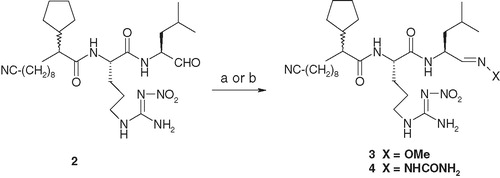
Table 1. Biological data for compounds.*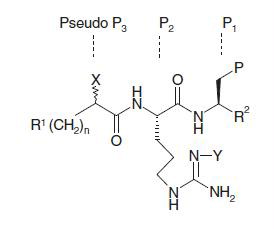 .
.
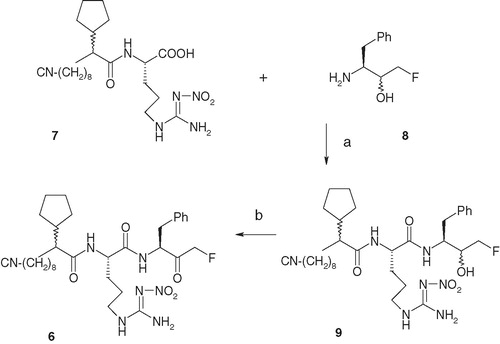
Compound 5 () was synthesized following a similar procedure that was utilized to synthesize parent compound 2 utilizing Fmoc-Arg(PMC) instead of Fmoc-Arg(NO2) in the originally reported schemeCitation16. Compound 6 () was accessed according to . Coupling of previously disclosed compounds 7Citation18 and 8Citation19 in the presence of peptide-coupling reagents BOP and HOBt generated compound 9 in 60% yield. Compound 9, on oxidation with Dess–Martin reagent, generated the target compound 6 in 70% yield.
Scheme 2. Reagents and conditions: (a) BOP, HOBt, NMM and DMF, 0 °C to room temperature, 2 h, 60%; (b) Dess–Martin reagent, CH2Cl2, 0 °C to room temperature, 2 h, 70%.
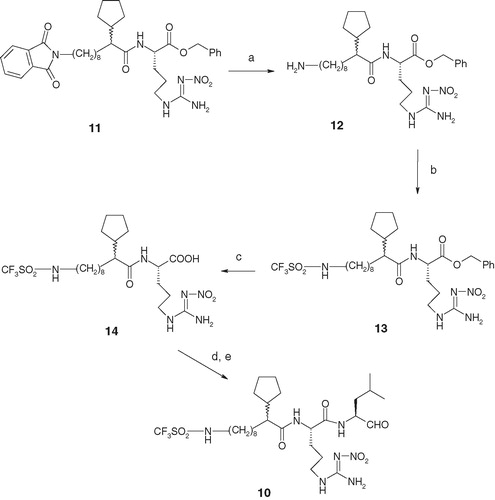
displayed the synthesis of target compound 10 (). Treatment of compound 11Citation18 with hydrazine generated compound 12 that was treated with trifluoromethylsulfonyl chloride in a basic media to generate sulfonamide 13; the yield was 60% over two steps. Removal of the benzyl group from compound 13 by catalytic hydrogenation generated compound 14 in 80% yield. Compound 14 was subsequently coupled with (S)-lucinol, in the presence of peptide-coupling reagents, to generate corresponding peptidomimetic alcohol (structure not shown) that on oxidation by the Dess–Martin reagent generated compound 10. Finally, Compounds 15–18 () were synthesized utilizing previously reported chemistryCitation16.
Scheme 3. Reagents and conditions: (a) hydrazine monohydrate, MeOH, heat, 1 h; (b) CF3SO2Cl, Et3N, CH2Cl2, −10 °C to room temperature, 2 h, 60% over two steps; (c) H2 gas, catalytic 10% Pd–C (50% water content), MeOH, 42–26 psi, 2 h, 80%; (d) (S)-leucinol, BOP, HOBt, NMM, DMF, 0 °C to room temperature, 2 h, 70%; and (e) Dess–Martin reagent, CH2Cl2, 0 °C to room temperature, 2 h, 80%.
Compounds were assayed for the CT-like activity of the human liver proteasome utilizing a fluorogenic substrate Succinyl-Leu-Leu-Val-Tyr-aminomethyl coumarinCitation16. Biological data of disclosed compounds are displayed in .
As mentioned, the proteasome is the first reported member of threonine proteinase class. Thus, exploration began with the mission of identifying a replacement for the carbonyl-derived aldehyde moiety known to inhibit serine and cysteine proteinases. As presented in , conversion of the aldehyde enzyme reactive group in compound 2 to corresponding imine-derivative 3 resulted in >100-fold loss in activity. However, activity was regained to some extent in hydrazine derivative 4, though ca 16 times less potent than compound 2. It is possible that the electrophilic nature of the carbon atom in –C=N– moiety in compound 4 was enhanced by the presence of electron-withdrawing carboxamide group attached to neighboring nitrogen atom. Replacement of the nitro group attached to the P2-ariginine moiety of compound 2 with PMC group (2,2,5,7,8-pentamethylchroman-6-sulfonyl) generated compound 5 that was ca threefold less potent than the parent compound, raising the possibility of steric constrain on activity from this site. Replacement of the aldehyde group of compound 2 with a fluoromethyl ketone group, known to inhibit a cysteine protease, e.g. calpain ICitation19, generated compound 6 that was significantly less active than parent compound 2 (however, note that P1 residue in this case was Phe). Replacement of the terminal nitrile group of compound 2 with a trifluoromethanesulfonamide moiety (compound 10) was not detrimental to the activity, reminiscent of similar tolerability of N-phthalimide moiety at that site in compound 1 (). In a similar fashion, compound 15 with a shortened chain length (n = 7), but containing a terminal caboxylate ester maintained the activity of compound 2. Replacement of the nitro group in compound 15 with PMC group generated compound 15 with a twofold drop in activity, establishing superiority of the nitro group. However, activity dropped eightfold when the cyclopentyl group at pseudo-P3 site in compound 15 was replaced by hydrogen (cf. activity of 17 versus 15). Interestingly, activity also dropped when the chain length was shortened from 7 to 6, even maintaining the cyclopentyl group at pseudo-P3 site (cf. compound 15 versus compound 18). Thus, it appeared that P3 region of this class of peptidomimetic inhibitors required an optimum balance of lipophilicity from the alkyl chain length and electronic contribution from a polar group.
In conclusion, in this Communication, we provided an account from our exploration in the area of inhibitors of CT-like activity of proteasome, a threonine proteinase. This study revealed that, in this class of inhibitors, while an aldehyde moiety can act as an enzyme reactive group, corresponding fluoromethylketone (active against cysteine proteinase calpain I) is not a suitable enzyme reactive group, thus differentiating these two classes of important proteinases. The research also highlighted the importance of an optimum balance of lipophilicity from an alkyl chain and electronic contribution from a polar group in the pseudo-P3 region of this class of peptidomimetic inhibitors. The knowledge gained from this study was instrumental in expanding the scope of the research that will be the basis of future publications.
Declaration of interest
Authors were employees of Cephalon, Inc., engaged in discovering novel proteasome inhibitors for the treatment of cancer.
Acknowledgements
Authors thank the members of the biology group for providing them with proteasome inhibition data. Authors wish to acknowledge Drs James C. Kauer and Jeffry Vaught for their keen interest, support and encouragement during the course of this study.
References
- Ciechanover A. Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Neurodegener Dis 2012;10:7–22
- Weissman AM, Shabek N, Ciechanover A. The predator becomes the prey: regulating the ubiquitin system by ubiquitylation and degradation. Nat Rev Mol Cell Biol 2011;12:605–20. Erratum in Nat Rev Mol Cell Biol 2011;12:686
- Lipkowitz S, Weismann AM. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat Rev Cancer 2011;11:629–43
- Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev 2007;107:687–717
- Nalepa G, Rolfe, M, Harper, JW. Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov 2006;5:596–613
- Lowe J, Stock D, Jap B, et al. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science 1995;268:533–9
- Groll M, Ditzel L, Lowe J, et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997;386:463–71
- Abu-Baker A, Messaed C, Laganiere Gaspar JC, et al. Involvement of the ubiquitin-proteasome pathway and molecular chaperones in oculopharyngeal muscular dystrophy. Hum Mol Genet 2003;12:2609–23
- Attaix D, Combaret L, Tilignac T, Taillandier D. Adaptation of the ubiquitin-proteasome proteolytic pathway in cancer cachexia. Mol Biol Rep 1999;26:77–82
- Chen M, von Mikecz A. Proteasomal processing of nuclear autoantigens in systemic autoimmunity. Autoimmun Rev 2005;4:117–22
- Kumatory Tanaka AK, Inamura N, et al. Abnormally high expression of proteasomes in human leukemic cells. PNAS 1990;87:7071–5
- Rolfe M, Chiu M, Pagano MI. The ubiquitin-mediated proteolytic pathway as a therapeutic area. J Mol Med 1997;75:5–17
- Sippl W, Collura V, Colland F. Ubiquitin-specific proteases as cancer drug targets. Future Oncol 2011;7:619–32
- Mitsiades CS, Mitsiades N, Hideshima T, et al. Proteasome inhibition as a new therapeutic principle in hematological malignancies. Curr Drug Targets 2006;7:1341–7
- Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol 2005;23:630–9
- Iqbal M, Chatterjee S, Kauer JC, et al. Potent inhibitors of proteasome. J Med Chem 1995;38:2276–7
- Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 1967;118:157–62
- Iqbal M, Diebold JL, Siman R, et al. Multicatalytic protease inhibitors. US Patent 1999; 5,990,083:1--48
- Chatterjee S, Ator MA, Bozyczko-Coyne D, et al. Synthesis and biological activity of a series of potent fluoromethyl ketone inhibitors of recombinant human calpain I. J Med Chem 1997;40:3820–8