
Abstract
A series of novel hybrid quinazoline–triazine derivatives was designed and synthesized from cyanuric chloride and anthranilic acid through sequential reactions, which contain different pharmacophores like quinazoline and substituted diaryl triazine (DATA) linked with ethylene diamine. All the newly synthesized compounds were characterized by infrared, 1H-NMR, 13C-NMR, MS and elemental analysis. Further, we evaluated the in vitro anti-HIV activity of the newly synthesized compounds against HIV-1 (IIIB) and HIV-2 (ROD) viral strains and as well as in vitro antimicrobial activity against four bacteria (Staphylococcus aureus, Bacillus cereus, Pseudomonas aeruginosa, Klebsiella pneumoniae) and two fungi (Aspergillus clavatus, Candida albicans) using the paper agar streak dilution method. The bioassay results indicate that four compounds namely 7d, 7n, 7r and 7s could be considered as possible potential agents.
Introduction
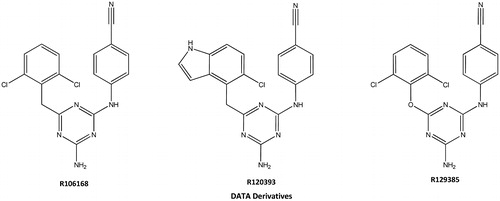
Since 2011, the acquired immunodeficiency syndrome (AIDS) had already claimed the lives of 1.7 million people, 2.5 million were newly infected with human immunodeficiency virus (HIV) and 34 million people are still living with HIVCitation1. Due to the prevalence of drug-resistant viral variantsCitation2, the highly active antiretroviral therapy (HAART) regimen plays a crucial role in the treatment of HIV-1 infection, typically involving concomitant treatment with a mixture of nucleoside and non-nucleoside HIV-1 RT (reverse transcriptase) inhibitors resulting in the combination of three to four drugs. However, drug resistance and side effects often compromise clinical treatmentCitation3. HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) have become an essential part of HAART, with unique antiviral high potency, low toxicity and exquisite selectivityCitation4. Among the NNRTIs, diaryl triazine (DATA) derivatives (), such as R129385, R120394 and R106168 displayed high potent activity against wild- and NNRTI-resistant strains of HIV-1 which has attracted considerable attention over the past few yearsCitation5.
Figure 1. The structures of DATA derivatives.
The development of hybrid molecules through the arrangement of dissimilar pharmacophores may provide compounds with attractive biological profilesCitation6. Several observations indicate that s-triazine analogues are the widely studied heterocyclic compounds owing to their broad spectrum of biological activity, including antimicrobialCitation7–10, antimycobacterialCitation11, anticancerCitation12,Citation13, antimalarialCitation14 and anti-HIV (NNRTIs) propertiesCitation15. The biological importance of quinazoline had already stimulated the medicinal chemists to consider it as a building block due to its broad range of pharmacological properties, such as antimicrobialCitation16–18, anti-HIV and anti-TMV (Tobacco Mosaic Virus)Citation19,Citation20, antitubercularCitation21, anticancerCitation22, anti-inflammatoryCitation23, anticonvulsantCitation24, antidepressantCitation25, hypolipidemicCitation26, antiulcerCitation27, analgesicCitation28 and immunotropic activitiesCitation29. Moreover, piperazine heterocycles are valuable structural units in drug researchCitation30–36.
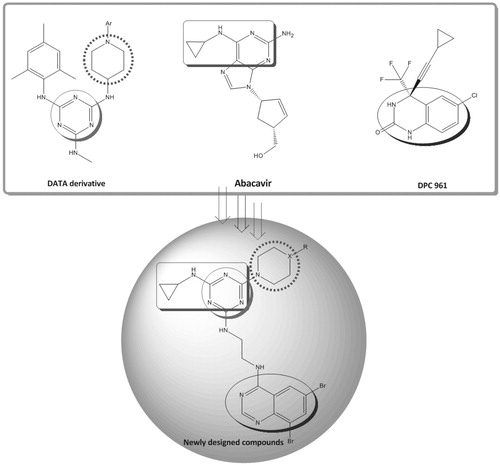
In context to the above rationale and in continuation of our ongoing program focused on finding new leads with anti-HIV activitiesCitation37–42, a novel hybrid series of DATA molecules with a combination of different pharmacophores such as triazine, quinazoline and piperazine that are structurally related to anti-HIV lead compounds, i.e. piperidine linked with DATA-NNRTI derivativesCitation43, abacavir having cyclopropyl amine groupCitation44 and DPC 961Citation4Citation5 with quinazoline moiety, are reported (). Herein, we synthesized a new series of ethylene diamine-linked quinazoline–triazine derivatives starting from the triazine ring of DATA.
Figure 2. The design of quinazoline–triazine hybrid derivatives.
Materials and methods
All experiments were conducted under scrupulously dry conditions and nitrogen atmosphere. All solvents were dried over an appropriate drying agent. Acetone was dried by heating under reflux over calcium hydride for several days followed by distillation. THF was dried by heating under reflux over sodium and benzophenone followed by distillation. 1,4-Dioxane, ethyl acetate, petroleum ether (fraction 50–70 °C), dichloromethane and methanol for chromatography were distilled before usage. Evaporation of solvents was carried out on a rotary evaporator under reduced pressure or using a high-vacuum pump. Analytical thin-layer chromatography was performed on Merck (Mumbai, India) precoated aluminum plates 60 F254 with a 0.2 mm layer of silica gel. NMR spectra were recorded on a 400 and 500 MHz spectrometer (Bruker AMX 400, Bruker AV 400, or a Bruker DRX 500; San Diego, CA) using DMSO as a solvent and TMS (Me4Si) as an internal standard. All 1H and 13C NMR chemical shifts are quoted in ppm and were calibrated on solvent signals. Multiplicities are given as s (singlet), d (doublet), dd (doublet–doublet), q (quadruplet), t (triplet) and m (multiplet). Liquid chromatography–mass spectrometry (LC/MS) analyses were performed using Shimadzu (Kyoto, Japan) LC-10AS pumps and a SPD-10AV UV-Vis detector (Shimadzu, Kyoto, Japan) with a Micromass Platform LC spectrometer (Mississippi, MS). LC/MS methods are detailed below. Preparative reverse phase (RP) HPLC was performed using two Shimadzu LC-8A pumps (Kyoto, Japan) and an SPD-10AV UV-Vis detector set at 220 nm on C18 RP columns (YMC Pack ODSA S5 20 × 100 mm or 30 × 250 mm; YMC Co. Ltd, Kyoto, Japan) eluting with methanol/water mixtures buffered with 0.1% trifluoroacetic acid.
3,5-Dibromo anthranilic acid (1)
Anthranilic acid (50 g, 0.36 mol) was dissolved and refluxed in glacial acetic acid (100 mL). Bromine (27.5 mL) was added slowly while reflux; colorless crystals took place. Then cooled to 25°C and filtered. The solid was rinsed with glacial acetic acid and then with benzene. The crude product was boiled up five times successively with 500 mL water, each filtration being made rapidly with suction. The insoluble residue consisting of the 3,5-dibromoanthranilic acid constituted two-thirds of the product. The pure acid was obtained by recrystallizing from alcohol: yield 102.2 g (95%).
6,8-Dibromoquinazolin-4(3H)-one (2)
A solution of 3,5-dibromo anthranilic acid (1) (5.00 g, 17 mmol) in formamide (15 mL, 0.29 mol) was heated slowly to 160 °C and maintained for 6 h. After the completion of reaction, the mixture after cooling was poured onto ice-water. The solid separated was filtered, dried and recrystallized from ethanol to give 6,8-dibromoquinazolin-4(3H)-one (2): yield 4.0 g (77%).
6,8-Dibromo-4-chloroquinazoline (3)
A mixture of 6,8-dibromoquinazolin-4(3H)-one (2) (5 g, 16.5 mmol), phosphorus oxychloride (10 mL, 39.8 mmol) and N,N-dimethyl aniline (10 mL, 86.3 mmol) was heated to 60–70 °C using a water bath for 2 h. After completion of the reaction, the excess phosphorus oxychloride was removed under reduced pressure, crushed ice (20 g) was added to the residue and adjusted to pH 7–7.5 by 1N NaOH solution. The separated solid was filtered, washed with water, dried and crystallized from dichloromethane affording the product 6,8-dibromo-4-chloroquinazoline (3): yield 5.0 g (94%).
N-(6,8-Dibromoquinazolin-4-yl)ethane-1,2-diamine (4)
A mixture of 6,8-dibromo-4-chloroquinazoline (3) (25 g, 77.6 mmol) and 1,2-diaminoethane (28 g, 25.27 mL, 0.4654 mol, 6 equiv.) was stirred under reflux for 8 h. After cooling to room temperature, the reaction was carried out under the water–ice mixture. The precipitate formed was filtered off, and the residue was washed with cold water. The compound was recrystallized from ethanol: yield 24.9 g (93%).
4,6-Dichloro-N-cyclopropyl-1,3,5-triazin-2-amine (5)
Cyanuric chloride (25 g, 0.14 mol) was suspended in chlorobenzene (125 mL) and cooled to −10 °C. Cyclopropylamine (8 g, 0.14 mol) was added slowly at −10 °C for 30 min. Then 30% of NaOH solution was added dropwise. The reaction mixture was stirred at −10 °C for 30 min and allowed to stand for a further 16 h at room temperature, after which it was washed with water (2 × 400 mL), dried over anhydrous sodium sulphate and filtered. The white crystals of cyclopropylamino-4,6-dichloro-s-triazine (5) were obtained through removal of the excess chlorobenzene by vacuum distillation: yield 26.7 g (96%).
4-Chloro-N-cyclopropyl-6-R-1,3,5-triazin-2-amine (6a–6s)
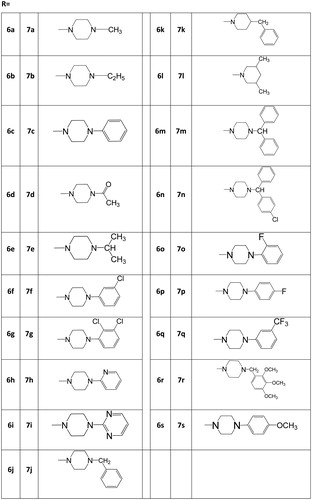
A solution of 4,6-dichloro-N-cyclopropyl-1,3,5-triazin-2-amine (5) (1.0 g, 4.9 mmol), relevant substituted piperazine or piperidine derivative (4.9 mmol) (presented in the table under ) and potassium carbonate (0.67 g, 4.9 mmol) in dry acetone (10 mL) was stirred for 5–6 h at room temperature. Progress of the reaction was monitored by TLC using solvent system toluene:acetone (8:2). After completion of the reaction, it was poured in the ice–water mixture, neutralized by dilute HCl and extracted with CHCl3, washed with water and dried over Na2SO4. The solution was concentrated under reduced pressure to afford the desire product (6a–6s).
Scheme 1. Overview of the synthetic strategy for the synthesis of N2-cyclopropyl-N4-(2-(6,8-dibromoquinazolin-4-ylamino)ethyl)-6-R-1,3,5-triazine-2,4-diamine (7a–7s).
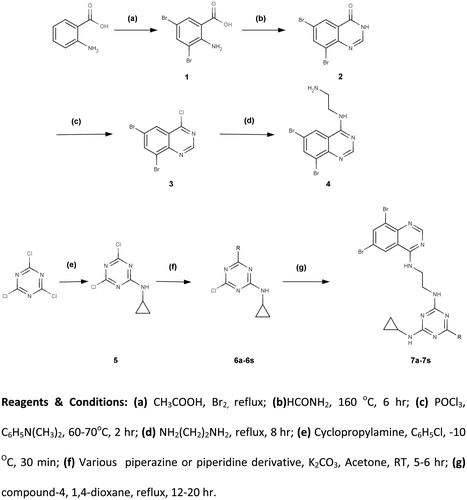
N2-cyclopropyl-N4-(2-(6,8-dibromoquinazolin-4-ylamino)ethyl)-6-R-1,3,5-triazine-2,4-diamine (7a–7s)
A solution of compounds (6a–6s) (1.0 mmol), N-(6,8-dibromoquinazolin-4-yl)ethane-1,2-diamine (4) (0.35 g, 1.0 mmol) and potassium carbonate (0.14 g, 1.0 mmol) in 1,4-dioxane (5 mL) was refluxed for 12–20 h. Progress of the reaction was monitored by TLC using toluene:acetone (7:3) as mobile phase. After completion of the reaction, the mixture was then treated with crushed ice and neutralized by dilute HCl. The precipitate thus obtained was filtered off, dried and recrystallized from THF to afford the desired compounds (7a–7s).
Medicinal chemistry part
In-vitro evaluation of antimicrobial activity
In order to study the antimicrobial properties of the novel hybrid quinazoline–triazine derivatives, several bacterial (Staphylococcus aureus MTCC 96, Bacillus cereus MTCC 430, Pseudomonas aeruginosa MTCC 741, Klebsiella pneumoniae MTCC 109) and fungal (Aspergillus clavatus MTCC 1323 and Candida albicans MTCC 183) species were selected and minimum inhibitory concentration (MIC) of the compound was determined by the agar streak dilution methodCitation46. A stock solution of the tested compound (200 μg/mL) in dimethylsulfoxide was prepared and graded quantities of the test compounds were incorporated in a specified quantity of molten sterile agar, i.e. nutrient agar for the evaluation of antibacterial and sabouraud dextrose agar for antifungal activity, respectively. The medium containing the test compound was poured into a petri dish at a depth of 4–5 mm and allowed to solidify under aseptic conditions. A suspension of the respective microorganism of approximately 105 CFU/mL was prepared and applied to plates with serially diluted compounds with concentrations in the range of 3.125–200 μg/mL in dimethylsulfoxide and incubated at (37 ± 1) °C for 24 h (bacteria) or 48 h (fungi). The lowest concentration of the substance that prevents the development of visible growth is considered to be the MIC value.
In vitro evaluation of anti-HIV assay
The evaluation of the anti-HIV activity of the synthesized compounds against HIV-1 strain (IIIB) and HIV-2 strain (ROD) in MT-4 cells was performed using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay method as earlier reportedCitation47,Citation48. In brief, stock solutions (10 times final concentration) of test compounds were added in 25 μL volumes to two series of triplicate wells so as to allow simultaneous evaluation of their effects in mock- and HIV-infected cells at the beginning of each experiment. Serial fivefold dilutions of test compounds were made directly in flat-bottomed 96-well microtiter trays using a Biomek 3000 robot (Beckman Instruments, Fullerton, CA). Untreated control HIV- and mock-infected cell samples were included for each sample. HIV-1(IIIB)Citation49 or HIV-2 (ROD)Citation50 stock (50 μL) at 100–300 CCID50 (50% cell culture infectious dose) or culture medium was added to either the infected or mock-infected wells of the microtiter tray. Mock-infected cells were used to evaluate the effects of the test compounds on uninfected cells in order to assess the cytotoxicity of the test compounds. Exponentially growing MT-4 cellsCitation51 were centrifuged for 5 min at 1000 rpm (220g) and the supernatant was discarded. The MT-4 cells were resuspended at a final concentration of 6 × 105 cells/mL and 50 μL volumes were transferred to the microtiter tray wells. After five days of incubation at 37 °C following infection, the viability of mock- and HIV-infected cells was examined spectrophotometrically by the MTT assay.
The MTT assay is based on the reduction of yellow-coloured MTT (Acros Organics, Geel, Belgium) by mitochondrial dehydrogenase of metabolically active cells to a blue–purple formazan that can be measured spectrophotometrically. The absorbances were read in an eight-channel computer-controlled photometer (Safire, Tecan), at two wavelengths (540 and 690 nm). All data were calculated using the median optical density value of three wells. The 50% cytotoxic concentration was defined as the concentration of the test compound that reduced the absorbance (optical density 540) of the mock-infected control sample by 50%. The concentration achieving 50% protection from the cytopathic effect of the virus in infected cells was defined as the 50% effective concentration. Anti-HIV activity and cytotoxicity of standard drug ddN/ddI were also performed by a similar method in MT-4 cells.
Results and discussion
In vitro anti-HIV assay
The novel synthesized compounds 6a–s and 7a–s were tested for their in vitro anti-HIV-1 (strain IIIB) and -HIV-2 (strain ROD) activity in human T-lymphocyte (MT-4) cells. Inhibitory concentrations (IC50) of synthesized compounds on HIV-1 and HIV-2 replications were measured by the inhibition of virus induced cytopathic effect in MT-4 cells. Cytotoxicity concentrations (CC50) of test compounds in mock infected MT-4 cells were also measured by the MTT-method. Anti-HIV activity and cytotoxicity of test compounds were compared with standard drug, 2H,3H-dideoxynucleosides/2′,3′-dideoxyinosine (ddN/ddI) against the replication of HIV-1 and HIV-2 in acutely infected MT-4 cells. The IC50 value of the synthesized molecules against HIV-1 and HIV-2 ranged from 2.22 to 113.45 μg/mL whereas the ddN/ddI inhibitory activities (IC50 4.35 and 6.96 μg/mL) were considered as standard. Cytotoxic concentration of test compounds ranged from 2.22 to 113.45 μg/mL whereas the standard drug dd/ddI showed an IC50 of 50 μg/mL in mock-infected MT-4 cells.
In the present study, some of the test compounds exhibited cytotoxic concentrations 2-fold higher than the standard drug ddN/ddI. Among the compounds tested, compounds 6d, 7g, 7h, 7n, 7p, and 7s with IC50 9.70, 2.22, 4.24, 9.56, 5.37 and 7.33 μg/mL, respectively, were more active against the replication of HIV-1 and HIV-2 in MT-4 cells. It should be noted that these IC50 values were inferior to the subtoxic concentrations. Besides, the above data showed no selectivity in anti-HIV activity since the selectivity index (SI = CC50/IC50) was below 1. Hence, the reported molecules were by themselves inactive, but may offer background information for active molecules. The results are presented in .
Table 1. Anti-HIV-1 and HIV-2 activity* (IC50 and CC50 in µg/mL) and cytotoxicity of compounds 6a–s and 7a–s in MT-4 cells.
Antimicrobial activity
The biological assay summarized in revealed that all the newly synthesized compounds 6a–s and 7a–s displayed biological activities vary with the differences in the functional group(s) or atom(s) attached to the piperazine moiety condensed to the nucleus. From the bioassay results it can be stated that all new derivatives showed appreciable antimicrobial activity. In the set of compounds 6a–s, compounds 6n with diphenyl substituent and 6s with methoxy substituent were most active against S. aureus and B. cereus bacteria, respectively, at 12.5 μg/mL of MIC. Compound 6d with the acetyl group to the piperazine ring exhibited moderate activity against three bacterial strains (B. cereus, P. aeruginosa and K. pneumoniae) at 50 μg/mL of MIC and also compound 6q with bearing trifluoromethyl substitution showed moderate activity against three bacteria (S. aureus, P. aeruginosa and K. pneumoniae) at 50 μg/mL of MIC. Analogues 6b, 6j, 6l and 6r were showed moderate activity against P. aeruginosa and K. pneumoniae at MIC 50 μg/mL. The antifungal results revealed that, the synthesized compounds showed variable degree of inhibition against the tested fungi. Acetyl substituted to piperazine derivative (6d), 2-fluoro phenyl group derivative (6o) and 2,3,4-trimethoxy benzyl derivative (6r) displayed higher activity against C. albicans as well as compound 6i with pyrimidyl ring against A. clavatus at an MIC of 25 μg/mL. Compound 6c with phenyl and compound 6q with trifluoromethyl-substituted phenyl to piperazine nucleus appeared with moderate to good inhibition for both the tested fungi (C. albicans and A. clavatus) at 50 μg/mL of MIC, whereas derivatives 6h, 6l, 6o and 6s appeared with moderate to good inhibition against A. clavatus and derivatives 6j and 6n against C. albicans at an MIC 50 μg/mL. While all the other derivatives in this set of compounds exerted poor activity profiles. In the set of compounds 7a–s, compounds 7d, 7n, 7r and 7s were potentially active against bacteria (S. aureus and B. cereus) at 6.25 μg/mL of MIC as well as against (P. aeruginosa and K. pneumonia) at 6.25 μg/mL and 12.5 μg/mL of MIC, respectively. Compound 7p with the 4-fluoro phenyl group appeared with significant inhibition of both Gram positive and Gram negative at 12.5 μg/mL of MIC, whereas compound 7c with phenyl substituent was effective against S. aureus at 12.5 μg/mL of MIC along with similar efficacy of analog 7p. Derivatives 7f and 7g with electron-withdrawing chloro-substituent had good activity against P. aeruginosa at an MIC level of 12.5 μg/mL, along with equal potency of compound 7s with electron-donating alkoxy substituent toward the same bacteria. Derivative 7n, 7r and 7s had lesser fungal activity against A. clavatus at 12.5 μg/mL, as well as against C. albicans at 25 μg/mL and 50 μg/mL of MIC, respectively, whereas compounds 7i with pyrimidine group and 7m with diphenyl substituent were also active against A. clavatus at an MIC of 12.5 μg/mL. Analogs 7f and 7g with a chloro-subtituent displayed good to moderate activity against A. fumigatus at an MIC of 25 μg/mL and 50 μg/mL of MIC, respectively. Compounds 5o and 5q with fluoro-substituent showed inhibition against C. albicans at an MIC of 25 μg/mL. Some analogs were found to exhibit moderate activity at >25 μg/mL of MIC; however, the activity level of many analogs was found to increase within the scaffolds studied in the research work presented here.
Table 2. In vitro antimicrobial activity in MIC* (μg/mL) of compounds 6a–s and 7a–s.
Conclusion
In this work, 38 novel ethylenediamine-linked quinazoline–triazine (DATA) derivatives were synthesized and examined thoroughly. The outcome showed that the type of group attached on the s-triazine ring may have a substantial impact on the biological activities of the aimed compounds. Out of the 38 compounds screened, compounds 7d, 7n, 7r and 7s exhibited promising in vitro antibacterial and antifungal effects. The derivatives were also evaluated for their in vitro anti-HIV-1 (strain IIIB) and HIV-2 (strain ROD) activity in human MT-4 cells. No specific anti-HIV activity was detected for any of the newly synthesized compounds. In general, the compounds showed improved antibacterial activity when compared to their antifungal activity. Among these compounds, a clear trend of improved activity has been shown to be due to the acetyl, dibenzyl, trimethoxy and mono-methoxy functionality at the nitrogen atom of piperazine bases condensed to the nucleus. In comparing the two set of compounds 6a–s and 7a–s, the incorporation of quinazoline moity linked to triazine via ethylene diamine is beneficial to antimicrobial activity. In brief, our findings might be helpful as leads for designing new molecule with potential bioactivities.
Declaration of interest
The authors have declared no conflict of interest.
Acknowledgements
Authors are very thankful to Prof. (Mrs) Shobhana K. Menon, Head, Department of Chemistry and Director, School of Sciences, Gujarat University, Ahmedabad, India, for her kind cooperation and providing support and research facilities. Mr Rahul Modh would like to acknowledge UGC New Delhi, for a Junior Research Fellowship and the Zydus Research Centre for providing spectral data.
Notes
Rahul Modh, Department of Chemistry, University School of Sciences, Gujarat University, Navrangpura, Ahmedabad 380 009, Gujarat, India. E-mail: [email protected]
Related Research Data
References
- UNAIDS 2011 World AIDS Day report. Available from: http://www.unaids.org. [last accessed 30 Oct 2012]
- Ragno R, Frasca S, Manetti F, et al. HIV-reverse transcriptase inhibition: inclusion of ligand-induced fit by cross-docking studies. J Med Chem 2004;48:200–12
- Hirschel B, Francioli P. Progress and problems in the fight against AIDS. N Engl J Med 1998;338:906–8
- De Clercq E. New developments in anti-HIV chemotherapy. Curr Med Chem 2001;8:1543–72
- Das K, Clark AD, Lewi PJ, et al. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J Med Chem 2004;47:2550–60
- Muregi FW, Ishih A. Next-generation antimalarial drugs: hybrid molecules as a new strategy in drug design. Drug Develop Res 2010;71:20–32
- Zhou C, Min J, Liu Z, et al. Synthesis and biological evaluation of novel 1,3,5-triazine derivatives as antimicrobial agents. Bioorg Med Chem Lett 2008;18:1308–11
- Srinivas K, Srinivas U, Bhanuprakash K, et al. Synthesis and antibacterial activity of various substituted s-triazines. Eur J Med Chem 2006;41:1240–6
- Modh RP, Patel AC, Mahajan DH, et al. Synthesis and evaluation of novel 4-substituted styryl quinazolines as potential antimicrobial agents. Arch Pharm 2012; 345:964--72
- Kaswala PB, Chikhalia KH, Shah NK, et al. Design, synthesis and antimicrobial evaluation of s-triazinyl urea and thiourea derivatives. Arkivoc 2009;11:326–35
- Machakanur SS, Patil BR, Pathan AH, et al. Synthesis, antimicrobial and antimycobacterial evaluation of star shaped hydrazones derived from 1,3,5-triazine. Der Pharma Chemica 2012;4:600–7
- Menicagli R, Samaritani S, Signore G, et al. In vitro cytotoxic activities of 2-alkyl-4,6-diheteroalkyl-1,3,5-triazines: new molecules in anticancer research. J Med Chem 2004;47:4649–52
- Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumour-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8
- Melato S, Prosperi D, Coghi P, et al. A combinatorial approach to 2,4,6-trisubstituted triazines with potent antimalarial activity: combining conventional synthesis and microwave-assistance. ChemMedChem 2008;3:873–6
- Xiong YZ, Chen FE, Balzarini J, et al. Non-nucleoside HIV-1 reverse transcriptase inhibitors. Part 11: structural modulations of diaryltriazines with potent anti-HIV activity. Eur J Med Chem 2008;43:1230–6
- Bartroli J, Turmo E, Algueró M, et al. New azole antifungals. 3. synthesis and antifungal activity of 3-substituted-4(3H)-quinazolinones1,2. J Med Chem 1998;41:1869–82
- El-Sharief A, Ammar Y, Zahran M, et al. Aminoacids in the synthesis of heterocyclic systems: the synthesis of triazinoquinazolinones, triazepinoquinazolinones and triazocinoquinazolinones of potential biological interest. Molecules 2001;6:267–78
- Modh RP, Patel AC, Chikhalia KH. Design, synthesis biological evaluation of some novel quinazolinone scaffolds. Med Chem 2012;8:182–92
- Purohit DM, Bhuva VR, Shah VH. Synthesis of 5-arylaminosulpho-N-acetylanthranilic acid, 6-arylaminosulpho-2-methyl-3-amino/3-N-chloroacetamido/3-N-arylaminoacetamido-4-(3H)-quinazolones as potential anti-HIV, anticancer and antimicrobial agents. Chem Indian J 2003;1:233–45
- Wang Z, Wang M, Yao X, et al. Design, synthesis and antiviral activity of novel quinazolinones. Eur J Med Chem 2012;53:275–82
- Murav'eva KM, Arkhangel'skaya NV, Shchukina MN, et al. Synthesis and tuberculostatic activity of aminoquinazoline derivatives. Pharm Chem J 1971;5:339–41
- Barker (ZENECA Limited) AJ, inventor; Quinazoline derivatives and their use as anti-cancer agents, European patent EP 0635498; 1995
- Laddha SS, Bhatnagar SP. A new therapeutic approach in Parkinson’s disease: some novel quinazoline derivatives as dual selective phosphodiesterase 1 inhibitors and anti-inflammatory agents. Bioorg Med Chem 2009;17:6796–802
- Aziza MA, Ibrahim MK, El-Helpy AG. Part II. Synthesis of novel-2-styryl-3-benzylidenimino-4(3H)-quinazolone derivatives of expected anticonvulsant activity. Al-Azhar J Pharm Sci 1994;14:193–201
- Jatav V, Mishra P, Kashaw S, Stables JP. CNS depressant and anticonvulsant activities of some novel 3-[5-substituted 1,3,4-thiadiazole-2-yl]-2-styryl quinazoline-4(3H)-ones. Eur J Med Chem 2008;43:1945–54
- Kurogi Y, Inoue Y, Tsutsumi K, et al. Synthesis and hypolipidemic activities of novel 2-[4-[(diethoxyphosphoryl)methyl]phenyl]quinazolines and 4(3H)-quinazolinones. J Med Chem 1996;39:1433–7
- Terashima K, Shimamura H, Kawase A, et al. Studies on antiulcer agents. IV: antiulcer effects of 2-benzylthio-5, 6, 7, 8-tetrahydro-4 (3H)-quinazolinones and related compounds. Chem Pharm Bull 1995;43:2021–3
- Alagarsamy V, Raja Solomon V, Dhanabal K. Synthesis and pharmacological evaluation of some 3-phenyl-2-substituted-3H-quinazolin-4-one as analgesic, anti-inflammatory agents. Bioorg Med Chem 2007;15:235–41
- Nawrocka W, Zimecki M. Syntheses of Novel 3-Amino-2(1H)-thioxo-4(3H)-quinazolinones and evaluation of their immunotropic activity. Part III. Arch Pharm 1997;330:399–405
- Upadhayaya RS, Vandavasi JK, Kardile RA, et al. Novel quinoline and naphthalene derivatives as potent antimycobacterial agents. Eur J Med Chem 2010;45:1854–67
- Al-Turkistani AA, Al-Deeb OA, El-Brollosy NR,El-Emam AA. Synthesis and antimicrobial activity of some novel 5-alkyl-6-substituted uracils and related derivatives. Molecules 2011;16:4764–74
- Ananda Kumar C, Vinaya K, Narendra Sharath Chandra J, et al. Synthesis and antimicrobial studies of novel 1-benzhydryl-piperazine sulfonamide and carboxamide derivatives. J Enzyme Inhib Med Chem 2008;23:462–9
- Upadhayaya RS, Sinha N, Jain S, et al. Optically active antifungal azoles: synthesis and antifungal activity of (2R,3S)-2-(2,4-difluorophenyl)-3-(5-{2-[4-aryl-piperazin-1-yl]-ethyl}-tetrazol-2-yl/1-yl)-1-[1,2,4]-triazol-1-yl-butan-2-ol. Bioorg Med Chem 2004;12:2225–38
- Banerjee D, Yogeeswari P, Bhat P, et al. Novel isatinyl thiosemicarbazones derivatives as potential molecule to combat HIV-TB co-infection. Eur J Med Chem 2011;46:106–21
- Singh KK, Joshi SC, Mathela CS. Synthesis and in vitro antibacterial activity of N-alkyl and N-aryl piperazine derivatives. Indian J Chem B 2011;50:196
- Kerns RJ, Rybak MJ, Kaatz GW, et al. Structural features of piperazinyl-linked ciprofloxacin dimers required for activity against drug-resistant strains of Staphylococcus aureus. Bioorg Med Chem Lett 2003;13:2109–12
- Patel RV, Kumari P, Rajani DP, et al. Antimicrobial, anti-TB, anticancer and anti-HIV evaluation of new s-triazine-based heterocycles. Future Med Chem 2012;4:1053–65
- Patel RB, Desai KR, Chikhalia KH. Synthesis and studies of novel homoveratryl based thiohydantoins as antibacterial as well as anti-HIV agents. Indian J Chem B 2006;45B:1716
- Patel RB, Chikhalia KH, Pannecouque C, Clercq E. Synthesis of novel PETT analogues: 3, 4-dimethoxy phenyl ethyl 1, 3, 5-triazinyl thiourea derivatives and their antibacterial and anti-HIV studies. J Braz Chem Soc 2007;18:312–21
- Patel RB, Chikhalia KH. Synthesis of heterocyclic and non-heterocyclic entities as antibacterial and anti-HIV agents. Indian J Chem B 2006;45B:1871
- Mahajan DH, Pannecouque C, De Clercq E, Chikhalia KH. Synthesis and studies of new 2-(coumarin-4-yloxy)-4, 6-(substituted)-s-triazine derivatives as potential anti-HIV agents. Arch Pharm 2009;342:281–90
- Desai SD, Desai KR, Chikhalia KH, et al. Synthesis of a novel class of some 1, 3, 5-triazine derivatives and their anti-HIV activity. Int J Drug Des Dis 2011;2:361–8
- Chen X, Zhan P, Liu X, et al. Design, synthesis, anti-HIV evaluation and molecular modeling of piperidine-linked amino-triazine derivatives as potent non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem 2012;20:3856–64
- Mallal S, Nolan D, Witt C, et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 2002;359:727–32
- Mutlib A, Chen H, Shockcor J, et al. Characterization of novel glutathione adducts of a non-nucleoside reverse transcriptase inhibitor, (s)-6-chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-3,4-dihydro-2(1h)-quinazolinone (dpc 961), in rats. possible formation of an oxirene metabolic intermediate from a disubstituted alkyne. Chem Res Toxicol 2000;13:775–84
- Hawkey P, Lewis DA. Medical bacteriology: a practical approach. Oxford, UK: Oxford University Press; 2004
- Baba M, Snoeck R, Pauwels R, De Clercq E. Sulfated polysaccharides are potent and selective inhibitors of various enveloped viruses, including herpes simplex virus, cytomegalovirus, vesicular stomatitis virus, human immunodeficiency virus. Antimicrob Agents Chemother 1988;32:1742–5
- Pannecouque C, Daelemans D, De Clercq E. Tetrazolium-based colorimetric assay for the detection of HIV replication inhibitors: revisited 20 years later. Nat Protoc 2008;3:427–34
- Popovic M, Sarngadharan MG, Read E, Gallo RC. Detection, isolation, continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science 1984;224:497–500
- Barré-Sinoussi F, Chermann JC, Rey F, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983;220:868–71
- Miyoshi I, Taguchi H, Kubonishi I, et al. Type C virus-producing cell lines derived from adult T cell leukemia. Gann Monogr 1982;28:219–28