
Abstract
Salmonella outer protein E, SopE, is a virulence factor that is secreted from gram-negative Salmonella enterica, which is pathogenic to humans and is responsible for gastroenteritis and typhoid fever. SopE targets the Rho GTPase family proteins of the host cell and manipulates them by mimicking GTPase regulatory protein guanine nucleotide exchange factor. The aim of this work is the investigation of novel inhibitors against SopE. Structure-based pharmacophore modeling was used to identify structural features that would be important for SopE recognition. Glide fragment library was used for four-point pharmacophore hypothesis development. Small-molecule database screening was performed based on a 3D similarity to the best pharmacophore hypothesis. Binding affinity of filtered database molecules to SopE was predicted quantitatively by molecular docking and scoring using Glide software. Top scoring hits were further analyzed and five molecules were proposed as potent and selective SopE inhibitors. Four out of five proposed molecules were found to be aminopurine derivatives.
Introduction
The Ras (Rat sarcoma) superfamily of GTPases consists of a large number of monomeric G-proteins, which are intracellular cell signaling proteins that take part in many diverse cellular functions, including growth, migration, adhesion, cytoskeletal integrity and survivalCitation1,Citation2. One subfamily is the Rho family of small (around 21 kDa) GTPases, which regulates actin dynamics and also participates in cell cycle progression, gene transcription, cell proliferation and motility, vesicular trafficking, phagocytosis and cytokinesisCitation3–6. Guanine nucleotide exchange factors (GEFs) reduce the affinity of Rho GTPases to guanosine diphosphate (GDP) by interacting with their switch regions and the nucleotide containing P-loopCitation6. Upon GEF binding, GDP is substituted by GTP, GTPase is activated and the downstream signaling cascade is turned onCitation7.
Salmonella genus are found widespread in cold- and warm-blooded animals and cause illnesses like typhoid fever and gastroenteritisCitation8. Salmonella outer effector proteins, which are secreted into the host cell by type III secretion system such as SopB, SopE and SopE2, modulate different cellular processes and are therefore involved in various stages of bacterial infection. SopE protein initiates bacterial internalization by membrane rufflingCitation8–10. SopE targets the Rho GTPases Rac1 and Cdc42. SopE acts as a GEF and stimulates nucleotide exchange from GDP to GTP, inducing signal transduction by the host Rho GTPasesCitation11. Bacterial uptake of SopE induces actin cytoskeleton rearrangement, which enables the entry of other effector proteins into the host cellCitation11.
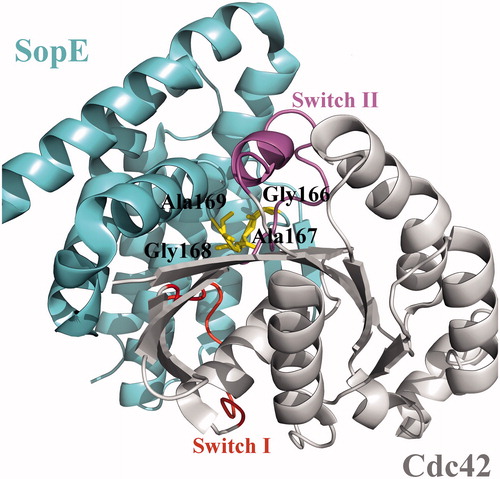
The structure of SopE consists of six α-helices in two, three-helix bundles that assume a Λ form and a two stranded β-sheet followed by an eight amino acid long loop (). This loop, which connects the two legs of the Λ shape, contains the catalytic core of SopE, a GAGA (residues Gly166, Ala167, Gly168, Ala169) motif (central loop in ). Catalytic core of SopE does not resemble the catalytic domain of other Rho-GEFs; whose structures have either DH-PH or DHR2 domainsCitation12. The crystal structure of the SopE–Cdc42 complex provides information about the interaction between these two proteins as well as clues about the mechanism by which the Cdc42 GTPase is activated by the SopE GEF. The structural analysis of the SopE–Cdc42 complex reveals that insertion of the GAGA loop between the nucleotide-binding regions of Cdc42 leads to major conformational changes in the Switch I (residues 28–40) and Switch II (residues 57–74) regions, resulting in reduced GDP affinityCitation11. Therefore, GAGA loop is crucial for bacterial GEF activity of SopE. Similar conformational changes in these switch regions have been observed by other eukaryotic GEF proteinsCitation11. Other than GAGA loop, multiple contacts are observed between Switch I region of Rho GTPase and Asp124 residue of SopE. Ile131 residue of SopE was also reported to have hydrophobic contacts with the Switch I region. Other reported Switch I interactions include Gln194 and Lys198 residues of SopE. GAGA loop residues Gly166, Gly168 and Ala169 interact with the Switch II region. Other Switch II contacting SopE residues are Asp103, Gln109, Ser165 and Thr174Citation11.
Figure 1. Crystal structure of the GEF domain of SopE in complex with Cdc42, shown in cartoon representation (PDB id: 1gzs:A,B). Switch regions of Cdc42 are shown in red and purple. Critical GAGA loop residues are shown in yellow.
Aminopurine is an organic fluorescent molecule that has been proven to be important in various fields and therefore may have a potential for new therapeutic purposes. It is extensively used in nucleic acid studies since it can function as both adenine and guanine analogCitation13,Citation14. Aminopurines were also found to be ATP competitive protein kinase inhibitors in many studiesCitation15–17. In this work, we employed a virtual screening (VS) approach that combines structure-based pharmacophore building, database filtering and molecular docking to identify small-drug like molecules that would inhibit GEF activity of SopE. Docking results were further analyzed according to their interactions with target, druglikeness and bioavailability. Up to date, no inhibitors for SopE have been reported. Here we computationally identified a number of aminopurine derivatives as novel potential inhibitors of SopE.
Methods
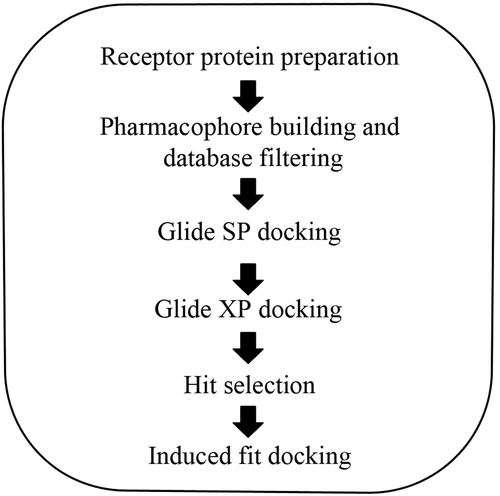
All VS applications were performed using Schrödinger Suite 2011 (LLC, New York, NY) on Linux platform (Linux Professional Institute, Sacramento, CA) using HP xw6600 Workstation (Hewlett-Packard Co, Palo Alto, CA). The following Schrödinger modules were used: Protein Preparation WizardCitation18, LigPrepCitation19, ConfGenCitation20, PhaseCitation21, QikPropCitation22, GlideCitation23–25 and Induced fitCitation26,Citation27. All modules were accessed via Maestro graphical interfaceCitation28 (all modules designed by LLC, New York, NY). The workflow depicted in is outlined in detail below.
Figure 2. Schematic workflow of VS and docking of SopE.
Database generation
The 3D structures of the small molecules from the big vendors of ZINC database (Table S1) were prepared via LigPrep and ConfGen scripts, and merged using Phase Database Generation module (LLC, New York, NY). The parameters used in the preparation step are the following: (a) possible ionization and tautomer states were generated at pH 7.0 ± 2.0; (b) chiralities were obtained from the 3D geometry of the structures; (c) for each compound, maximum four low energy isomers, one ring conformation and 10 conformations per rotatable bond were generated; (d) 50 steps of energy minimization were carried out for the conformers with the truncated Newton (TCNG) method using the OPLS_2005 force field and a distance dependent dielectric constant of 4; (e) conformers with high-energy ionization/tautomer states were automatically removed and number of conformers was limited to 100 per compound, which is the default value for conformer generation in Phase Database Generation module; (f) a built-in QikProp script predicted ADME and druglikeness properties of the molecules; (g) molecules that violated Lipinski’s rule or had reactive functional groups were removed. Database preparation yielded a total of 25.8 million conformations from the initial 500 000 molecules.
Receptor protein preparation
Coordinates of the target protein SopE (PDB id: 1gzs) were obtained from RCSB Protein Data Bank (NJ)Citation29. One SopE chain from the crystal structure, which contains the SopE–Cdc42 in dimeric form, was used to prepare the protein within the Protein Preparation Wizard. Bond orders were assigned, hydrogens were added to the heavy atoms and all waters were deleted. Reorientation of certain hydroxyl and thiol groups, amide groups of asparagines, glutamines and imidazole ring of histidines, protonation states of histidines, aspartic acids and glutamic acids were optimized at neutral pH. Using force field OPLS_2005 (NY), minimization was carried out setting maximum heavy atom RMSD to 0.30 Å.
Receptor grid generation
After preparation, receptor grids were generated with Glide by specifying the binding site with a 3D cubic box. Enclosing a box of 20 Å side lengths was placed by centering the critical GAGA loop. Potential energies of receptor atoms confined in this box were calculated without scaling their van der Waals radius. Gly166, Gly168 and Ala169, three GAGA loop residues that are known to interact with Cdc42, were identified as hydrogen bond partners for later use as constraints during docking. Rotation of hydroxyl groups was not allowed.
Structure-based pharmacophore generation
The database, which comprised 500 000 molecules, was filtered using a pharmacophore hypothesis in order to optimize the docking and screening time. Due to the fact that there are no previously identified ligands for SopE; a method that couples structure information with pharmacophore building method, which is called E-pharmacophores (E-pharm), was employed. This methodology makes use of the receptor binding site structural information, and generates pharmacophore sites based on the protein residues that can make interactions upon ligand binding. In order to identify such residues, E-pharm procedure required an initial docking stage. A set of 667 unique small (6–37 atoms, MW range 32–226) fragmentsCitation30, derived from the molecules in the medicinal chemistry literature (by Schrödinger Company), were used as ligands. The interface between SopE and Cdc42 in the complex structure was examined in detail in order to identify key interacting residues. The contribution of the GAGA loop to Cdc42 recognition has been reported in the literatureCitation11. Extra precision (XP) docking without constraints was performed using all fragments and the GAGA loop of SopE as a binding site. XP descriptors were generated to obtain atom-level energy terms such as hydrogen bond interaction, electrostatic interaction, hydrophobic enclosure and π–π stacking interaction. E-pharm script mapped XP energetic terms onto each fragment atom. Pharmacophore sites were determined on fragment atoms with Phase, using the built-in set of six features: hydrogen bond acceptor (A), hydrogen bond donor (D), hydrophobic (H), negative ionizable (N), positive ionizable (P) and aromatic ring (R) site. Energetic contribution of each pharmacophore feature was quantized by assembling energies of the atoms that comprise pharmacophore site. Minimum distance between site points was kept as 2.0 Å and minimum distance between site points with same feature was kept as 4.0 Å. Pharmacophore sites were ranked based on their summed energies, and top ranking sites were combined to create a four-point hypothesis. Acceptor-acceptor-donor-negative ionizable (AADN) hypothesis was selected and the database was pre-filtered based on their 3D similarity to the selected pharmacophore hypothesis, allowing the minimum fitness value to be 0.
Glide docking
A total of 9970 molecules obtained by filtering were used for receptor docking. Glide standard precision (SP) docking was performed with 9970 molecules and top 10% hits based on GlideScore were re-docked to SopE in XP mode, keeping all docking parameters as default. At least one hydrogen bond defined on GAGA loop residues was selected to be satisfied. Using Monte Carlo random search algorithm, ligand poses were generated for each input molecule, and binding affinity of these molecules to SopE was predicted with GlideScore. Potential energies of the docked molecules were also predicted with empirical E-model scoring function. Post-docking minimization was performed with OPLS_2005 force field and at most five poses per ligand were saved. Strain energies of bound and free forms of ligands were calculated and hits having more than 4 kcal/mol energy difference between the two forms received a penalty equal to the quarter of their strain energy difference, which is added to docking score.
Induced fit docking
Induced fit docking (IFD), which allows both receptor side chain and ligand flexibility were carried out with proposed molecules with an inhibition potential towards SopE. IFD was performed to determine the interaction potential of proposed molecules to SopE when receptor-side chain flexibility is induced and no constraints are given. Receptor grid parameters were kept same as Glide docking, except no bonding constraints were determined on GAGA loop residues. Other internal settings and parameters in induced fit workflow were kept at their default states and values.
Results and discussion
In order to find potent inhibitors against SopE, structure-based VS was combined with pre-filtering of small molecule database with pharmacophore building. In this work, a hybrid screening method that combines pharmacophore building and molecular docking was performed to improve screening efficiency. Phamacophore modeling was employed to make use of the binding site shape, size and chemical properties in filtering the database. This allowed us to start the workflow with a larger database, which was then reduced by pharmacophoric search, and the likelihood of finding good-binding ligands within a reasonable screening time could be enhanced. Filtered molecules were docked to receptor SopE in multistep Glide docking protocol and potential inhibitors were identified. A simplified workflow of the VS methodology is presented in .
Structure-based pharmacophores
Ligand-based pharmacophore building, which facilitates the database filtering and screening process, requires an initial set of known ligands. However, there are no known ligands identified for SopE to date. Instead, knowledge about the receptor itself was utilized for determining the necessary structural and chemical features in a potential ligand for receptor binding. Fragments were docked to the binding site of the target SopE, near the GAGA loop, as explained in “Methods” section. Docking calculations were performed and protein-fragment interactions were characterized based on energetic contributions of each site on the fragments to the docking score. Thus, coordinates and types of pharmacophore sites (e.g. hydrogen donor, acceptor) that show good binding to GAGA loop were determined, and these sites were ranked.
A total of 21 pharmacophore sites that significantly contribute to the docking score were detected. shows all fragment-binding modes that were docked to SopE-binding site. Ranking of all possible pharmacophore sites as well as energetic contributions, coordinates and feature types are listed in and are shown in . In order to create a pharmacophore hypothesis, E-pharm provides two options: (a) allowing the user to select the sites that will constitute the hypothesis and (b) clustering based on site–site distances and total number of site points, specified by the user. Phase allows the number of pharmacophore site points to be between three and sevenCitation31. This value was selected as four, because when more site points were included, the likelihood of finding a cluster was decreased. In addition, hypothesis with more site points would yield fewer matches in database, and possibly eliminating potential ligands. The 21 pharmacophore sites were clustered and four high ranking site points that are within 4.2 Å–7.5 Å to each other were determined by E-pharm, to create the hypothesis AADN based on site points A708, N1548, A42 and D1091 (). The angle and distances between the site points are given in Table S2 and Figure S1. D995, which has the highest rank, was not picked because it is at least 10 Å away from the four site points. The AADN hypothesis was superimposed on the Cdc42–SopE complex and the residues that would potentially be replaced by the site points which would prevent Cdc42–SopE interaction were identified. A708 was located near Ala169. Likewise, N1548 site was located near Lys198SopE, possibly preventing its interaction with Asp38Cdc42. A42 was occupied by Val36Cdc42 whereas D1091 was in the vicinity of Thr35Cdc42. Both residues are in Switch I region of Cdc42 and they were known to interact with Asp124SopECitation11.
Figure 3. (A) Binding modes of all fragments superimposed at the binding site of SopE in stick representation. (B) Pharmacophore site points as spheres located on the binding site of SopE, determined by E-pharm. Top six sites based on energetic contribution are labeled. All site points are colored and labeled according to their feature type (donor, acceptor, hydrophobic or ring).
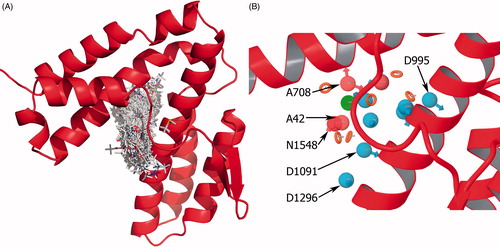
Table 1. Pharmacophore features and ranks by E-pharm. Numbers in feature labels are indices which are automatically assigned during feature generation.
Molecular docking
Pharmacophore pre-filtering with the four site point E-pharm hypothesis AADN yielded 9970 compounds from the small molecule database generated from ZINC. The molecules obtained by filtering with AADN hypothesis were docked to SopE with Glide software to predict binding affinity of molecules to SopE, and to investigate ligand–protein interactions.
The SopE coordinates used in this study were taken from the crystal structure of the SopE–Cdc42 complex. The structure of SopE in its apo form is not available, but there are two homologous proteins, BopECitation32 and SopE2Citation33, for which structure determination and relaxation measurements by NMR have been performed. These studies show that the GAGA loop is a flexible structural element. This is an expected result since this loop is a connector between the two arms of the Λ shape and comprises small and flexible residues. The Λ shape is suggested to assume open and closed conformations, in which the open conformation can bind Cdc42 but the closed conformation assumed by BopE would be in steric clash with Cdc42. In our work, the flexibility of the GAGA loop was taken into account by choosing to use the open conformation assumed by SopE in the SopE–Cdc42 complex. Even though only one conformation was used, this conformation allowed the maximum space for docking.
The position and orientation of each ligand relative to the receptor protein was determined and scored with internal scoring function GlideScore. Glide SP and XP docking modes were used, successively. At least one hydrogen bond with Gly166, Gly168 and Ala169 was set as constraint. Four hundred and eighty-four compounds satisfied the constraint criterion and were docked in Glide XP mode. GlideScores of these 484 compounds vary between −8.2 and −0.4 kcal/mol in Glide XP mode. Detailed analysis of docking parameters was carried out for hits with GlideScore −6.5 kcal/mol or higher. This threshold yielded 20 hits out of 484 compounds. 2D structures of all compounds are listed in Table S3.
Molecules were sorted according to their GlideScore values in . Detailed GlideScore components of each molecule are listed in Table S4. Emodel scoring function (, last column) includes penalty terms for internal strain energy of the generated poses. Therefore, GlideScore was used for comparing poses of different hits, and Emodel was used to decide between different conformers (or poses) of a particular ligand. Five molecules out of final 20 hits were selected as potent inhibitors of SopE based on the GlideScore and Emodel as well as additional criteria, such as absorption, distribution, metabolism and excretion (ADME) considerations and druglikeness of ligands and strain energy differences of ligands.
Table 2. Docking results of top 20 hits in kcal/mol. ZINC IDs and strain corrected GlideScores for proposed inhibitors are indicated by (*).
Pharmacodynamic properties, oral bioavailability and druglikeness as well as other molecular descriptors for the 20 candidate hits were determined by QikProp (LLC, New York, NY; Table S5). ZINC03860635 and ZINC03874923 have poor predicted human oral absorption (HOA) value, at 0.7% and 0.1%, respectively. ZINC00375097 was also observed to lack hydrophobic solvent accessible surface area with 0 Å2. Therefore, these three molecules were eliminated. Highest predicted HOA was observed for ZINC17076964 at 86.4%. ZINC19910989 and ZINC17076964 showed the highest lipophilicity at 3.231 and 3.433, respectively.
Docking allows ligands to be strained, so that they can fit better into the binding site. Since too much strain may indicate false positives, energies of free and docked conformations of hits were calculated and strain penalties were added to docking score. As a result, 11 out of 20 hits received strain penalties. ZINC10274669 had the highest strain penalty with 7 kcal/mol, which is almost as much as its original docking score. ZINC03860635, ZINC03874923 and 9th ranking ZINC05033974 also received relatively high strain penalties, which lowered their GlideScores considerably.
Molecules with acceptable ADME properties and GlideScore above −7 kcal/mol after strain correction were identified as ZINC00370772, ZINC09214236, ZINC17020721, ZINC19910989 and ZINC05033974. ZINC09214236, ZINC19910989 and ZINC05033974 had multiple poses. Selection of poses of a particular molecule was performed mainly based on their Emodel values. If Emodel value difference was not significant, docking scores were also considered. Poses with too much strain were also eliminated. Accordingly, 10th ranking pose of ZINC09214236, 8th ranking pose of ZINC19910989 and 12th ranking pose of ZINC05033974 were selected. Proposed SopE inhibitors are indicated by star in , and their ADME properties are given in .
Table 3. ADME and druglikeness properties of the selected ligands by QikProp.
Structures of final 20 conformations were analyzed and clustered by their 2D structural similarities via ChemMine Web Tool (CA) (http://chemmine.ucr.edu/). Hierarchy of clusters based on pairwise compound similarities were defined using the Atom Pair descriptors and Tanimoto coefficientCitation34. The similarity tree output is provided in Figure S2. The results show that these molecules form two clusters, with one cluster of five compounds and the other cluster with the rest of the compounds. The most significant structural dissimilarity between these clusters was that, the molecules in the first cluster contain an aminopurine ring while molecules in the second cluster do not contain an aminopurine ring. It was observed that four top ranking compounds that were proposed as SopE inhibitors belong to the first cluster.
Binding mode and interaction analysis of the selected hits
Superposition of the proposed molecules based on their interaction with SopE showed that their binding modes are similar (). Proposed molecules with aminopurine groups overlaid well and similar spatial arrangement of S1, S2 and S4 was observed. In contrast, aminopurine group of S5 was faced toward GAGA loop (). The five selected compounds satisfied at least two out of four pharmacophore sites with a 3 Å tolerance, suggesting that the pharmacophore building and docking results converged. The superposition of the selected compounds on the pharmacophore AADN hypothesis and the distances between the pharmacophore sites and the respective ligand atoms are given in Table S6. The interactions of the five proposed inhibitors were analyzed to verify that the ligands made contacts with previously identified important residues of SopE. The binding mode of selected molecules along with receptor hydrogen bond interactions are represented in . The list of hydrogen bond interactions between molecules and SopE is given in .
Figure 4. (A)–(E) Glide XP binding modes of proposed molecules S1–S5 at SopE binding site. SopE is shown in ribbon representation. Ligands and SopE residues interacting with the ligand are shown in stick representation, colored by atom type. Hydrogen bonds between ligands and SopE are indicated by black dashed lines. (F) Binding mode overlay of five proposed molecules. SopE binding site is in surface representation and ligands are in stick representation, colored by molecule.
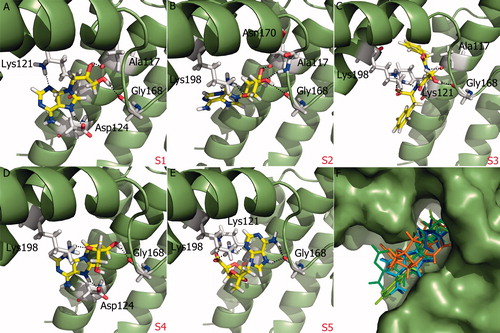
Table 4. Summary of H-bond interactions between and proposed molecules of SopE. “+” sign indicates presence of a hydrogen bond and number of signs indicates the number of hydrogen bonds between ligand and a particular SopE residue.
All proposed molecules favored the interaction with Gly168 residue of the GAGA loop, which is reported to be an important promoter of nucleotide release. Mutation of Gly168 to alanine was shown to abolish SopE-mediated nucleotide releaseCitation11. Therefore, Gly168 interactions were interpreted as a positive outcome. It was observed that Gly168 residue preferred interactions with either hydroxyl group or amine group in ring systems. It should be noted that structure-based VS of SopE was carried out with at least hydrogen bond criterion on GAGA loop residues Gly166, Gly168 and Ala169. Asp124 and Lys198 were also reported to be important residues for stabilizing Switch I regions of small GTPases. Lys198 interactions were mostly with lone oxygen or nitrogen atoms of ligands as well as nitrogen atoms. Two interactions between charged –NH groups and Asp124 residue of SopE were observed. Ala117 residue also participated in hydrophobic interactions. Four out of five proposed SopE inhibitors include an aminopurine ring, which may be a functionally important group upon SopE inhibition. These aminopurine rings bind to Lys 121, Asp124, Gly168 or Lys198 residues of SopE. One of the proposed molecules, S1 or ZINC00370772 is used in the therapy of viral diseases. This molecule was denoted as a novel nucleoside analog and reported to inhibit the replication of several DNA and RNA viruses, including vaccinia, herpes simplex (types 1 and 2), measles and vesicular stomatitisCitation35.
Induced fit docking
IFD was carried out with the proposed molecules, in order to study their binding poses, interactions with the receptor and predicted binding affinity in terms of docking score, when both receptor-side chain and ligand atoms were flexible. IFD binding modes along with their receptor interactions are given in and IFD scores are listed in .
Figure 5. (A)–(E) IFD binding modes of proposed molecules S1–S5 at SopE binding site. SopE is shown in ribbon representation. Ligands and SopE residues interacting with the ligand are shown in stick representation, colored by atom type. Hydrogen bonds between ligands and SopE are indicated by black dashed lines. (F) Binding mode overlay of five proposed molecules. SopE binding site is in surface representation and ligands are in stick representation, colored by the molecule.
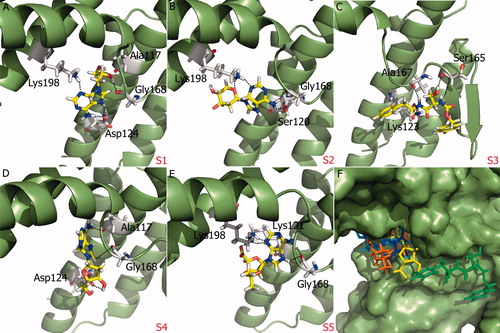
Table 5. IFD Score of proposed molecules in kcal/mol. Docking score of the best pose is presented for each ligand.
In IFD, the highest scoring binding mode of S1 was essentially similar to its Glide XP binding mode (). S1 was able to interact with Ala117, Asp124 and Gly168, as in Glide docking. The Lys121 interaction with the aminopurine ring was not observed in IFD. Instead, Lys198 made a hydrogen bond with S1. Highest scoring binding mode of S2 was also similar to its original Glide XP binding mode in which it was able to interact with Asn170 and Lys198. However interactions with Gly168 were not observed. In the second ranking binding mode of this ligand, which has a completely different orientation (IFD score: −7.18 kcal/mol), the aminopurine ring was able to interact with Ser120, Gly168 and Lys198 (). The highest scoring docking mode of S3 generated in IFD protocol had a completely different binding mode than its original binding mode, in which the molecule showed interactions with Lys123, Ser165 and Ala167 (). This ligand also showed the lowest score in IFD (). Investigation of the other poses of S3 showed that in all orientations, this ligand preferred to interact with Ala167 of the GAGA loop instead of Gly168. Highest scoring pose of S4, which is considerably different from its original binding pose, was able to interact with Ala117, Asp124 and Gly168 (). IFD binding mode of S5 with highest score was similar to its original docking orientation, interacting with Lys121, Gly168 and Lys198 (). shows the overlay of IFD binding modes of proposed molecules. All molecules except S3 had similar binding modes. Overall, induced fit results showed that identified interactions between proposed molecules with aminopurine ring (S1, S2, S4, S5) and SopE in Glide docking were also observed when receptor was flexible, while S3 maintained its contact with the GAGA loop by forming interactions with residue Ala167, different from other ligands.
Selectivity
Binding selectivity of proposed molecules to SopE homologs, which are SopE2 (Pdb id: 1e6e) with 70% amino acid sequence identityCitation36–39 and BopE (Pdb id: 2jok) with 27% amino acid sequence identity to SopECitation40 were investigated. The catalytic GAGA motif of SopE is conserved in SopE2 (residues 166–169) whereas GAGT motif is present in BopE (residues 171–174)Citation32. Glide XP docking with strain correction was performed on these proteins centering GAGA/GAGT motif around a 20 Å grid box. Proposed molecules were docked to SopE2 and BopE active site with the same docking parameters used for SopE, including at least one hydrogen bond constraint on GAGA/GAGT motif. Since GAGA loop is critical for the GEF function of these proteins, hydrogen bond interaction between proposed ligands and GAGA/GAGT loop was considered as an indicator for potential inhibitory action. No ligand pose could be generated for BopE, indicating that the proposed SopE molecules are not able to bind to BopE with the specified constraints. On the other hand, S1, S3 and S5 were successfully docked to SopE2 satisfying the GAGA motif H-bond constraint. The selectivity docking results are listed in . None of the hits received strain penalty, and the strain adjusted GlideScores are between −4.58 and −2.92, which is considerably lower than SopE docking results (around −7.50 kcal/mol). However, despite their lower docking scores, hits were able to interact with Gly166 and Ala169. These results suggest that the proposed molecules are selective toward SopE in terms of docking score, but also have inhibition potential against SopE2.
Table 6. Docking results of proposed SopE inhibitors to SopE2 in kcal/mol.
Conclusions
Potent inhibitors of SopE were investigated with computer-aided drug discovery tools. Ligand-based and structure-based VSapproaches were used employing the Schrödinger Suite 2011 modules. Database molecules were screened and pre-filtered, according to their 3D similarity to the selected pharmacophore AADN hypothesis. Filtered database molecules were docked to receptors in two successive docking modes, allowing ligand flexibility. Binding poses of input molecules were determined and scored with GlideScore. Top 20 molecules were subjected to a number of post-docking analyses, including strain energy calculation, assessment of ADME and druglikeness properties. Ultimately, five potential SopE inhibitors molecules with GlideScores around −7.5 kcal/mol and acceptable pharmacodynamic properties were proposed.
The aim of this VS study was to find a potential SopE inhibitor that would hinder the interaction between SopE and Cdc42. The inhibitor binding site was selected such that the inhibitor would interact with the GAGA loop residues that contact the switch regions of Cdc42. Detailed analysis of the interactions of the molecules with SopE showed that the molecules made contacts with Gly168 of the GAGA loop, which interacts with the Switch II of the GTPase Cdc42, and does not stimulate nucleotide exchange on Cdc42 when mutated to alanine. Four out of five proposed molecules were observed to have an aminopurine ring, which may be a functional group for SopE inhibition and needs further investigation in biological assays. Ligand–receptor interactions were investigated with more accurate IFD, and molecules with the aminopurine ring showed favorable interactions, which again points out the importance of this group. Additionally, selectivity of these molecules to known SopE homologs SopE2 and BopE were studied. None of the proposed molecules were able to interact with GAGT motif of BopE whereas three molecules (S1, S3 and S5) interacted with GAGA motif of SopE2 with considerably lower docking scores (−4.5 kcal/mol or less) compared to SopE, implying that proposed molecules are selective toward SopE but have a possibility to inhibit homologous SopE2 GEF as well.
Declaration of interest
The authors report no declarations of interest.
This work is financially supported by the Scientific and Technological Research Council of Turkey (TÜBİTAK) through project 111M444. The authors thank Prof. Sumru Ozkirimli for insightful discussions and her valuable contributions.
References
- Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev 2001;81:153–208
- Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci 2005;118:843–6
- Toksoz D, Merdek KD. The Rho small GTPase: functions in health and disease. Histol Histopathol 2002;17:915–27
- Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol 2004;265:23–32
- Evers EE, Zondag GC, Malliri A, et al. Rho family proteins in cell adhesion and cell migration. Eur J Cancer 2000;36:1269–74
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature 2002;420:629–35
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 2005;6:167–80
- Grassl GA, Finlay BB. Pathogenesis of enteric Salmonella infections. Curr Opin Gastroenterol 2008;24:22–6
- Layton AN, Galyov EE. Salmonella-induced enteritis: molecular pathogenesis and therapeutic implications. Expert Rev Mol Med 2007;9:1–17
- Jones MA, Hulme SD, Barrow PA, Wigley P. The Salmonella pathogenicity island 1 and Salmonella pathogenicity island 2 type III secretion systems play a major role in pathogenesis of systemic disease and gastrointestinal tract colonization of Salmonella enterica serovar Typhimurium in the chicken. Avian Pathol 2007;36:199–203
- Buchwald G, Friebel A, Galan JE, et al. Structural basis for the reversible activation of a Rho protein by the bacterial toxin SopE. EMBO J 2002;21:3286–95
- Boyle EC, Brown NF, Finlay BB. Salmonella enterica serovar Typhimurium effectors SopB, SopE, SopE2 and SipA disrupt tight junction structure and function. Cell Microbiol 2006;8:1946–57
- Frederiksen S. Effect of 2-aminopurine and 2-aminopurine 2′-deoxyriboside on nucleic acid synthesis in Ehrlich ascites cells in vitro. Biochem Pharmacol 1965;14:651–60
- Elion GB, Hitchings GH. Antagonists of nucleic acid derivatives. IV. Reversal studies with 2-aminopurine and 2,6-diaminopurine. J Biol Chem 1950;187:511–22
- Metheny LJ, Nelson DA, Ludlow JW. Effect of the kinase inhibitor 2-aminopurine on the cell cycle stage-dependent phosphorylation state of the retinoblastoma protein. Exp Cell Res 1995;218:151–4
- Xiang J, Lahti JM, Kidd VJ. 2-Aminopurine overrides a late telophase delay created by ectopic expression of the PITSLRE beta 1 protein kinase. Biochem Biophys Res Commun 1994;199:1167–73
- Hu Y, Conway TW. 2-Aminopurine inhibits the double-stranded RNA-dependent protein kinase both in vitro and in vivo. J Interferon Res 1993;13:323–8
- Schrödinger Suite 2011 Schrödinger Suite; Epik version 2.2, Schrödinger, LLC, New York, NY, 2011; Impact version 5.7, Schrödinger, LLC, New York, NY, 2011; Prime version 2.3, Schrödinger, LLC, New York, NY; 2011
- LigPrep, version 2.5, Schrödinger, LLC, New York, NY; 2011
- ConfGen, version 2.3, Schrödinger, LLC, New York, NY; 2011
- Phase, version 3.3, Schrödinger, LLC, New York, NY; 2011
- QikProp, version 3.4, Schrödinger, LLC, New York, NY; 2011
- Halgren TA, Murphy RB, Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 2004;47:1750–9
- Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004;47:1739–49
- Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 2006;49:6177–96
- Sherman W, Day T, Jacobson MP, et al. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 2006;49:534–53
- Sherman W, Beard HS, Farid R. Use of an induced fit receptor structure in virtual screening. Chem Biol Drug Des 2006;67:83–4
- Maestro, version 9.2, Schrödinger, LLC, New York, NY; 2011
- Bernstein FC, Koetzle TF, Williams GJ, et al. The Protein Data Bank. A computer-based archival file for macromolecular structures. Eur J Biochem 1977;80:319–24
- Schrödinger Fragment Library (2009) Schrödinger, Inc. Available from: http://www.schrodinger.com/productpage/14/5/73/ [last accessed 12 Apr 2012]
- Dixon SL, Smondyrev AM, Knoll EH, et al. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J Comput Aided Mol Des 2006;20:647–71
- Upadhyay A, Wu HL, Williams C, et al. The guanine-nucleotide-exchange factor BopE from Burkholderia pseudomallei adopts a compact version of the Salmonella SopE/SopE2 fold and undergoes a closed-to-open conformational change upon interaction with Cdc42. Biochem J 2008;411:485–93
- Williams C, Galyov EE, Bagby S. solution structure, backbone dynamics, and interaction with Cdc42 of Salmonella guanine nucleotide exchange factor SopE2. Biochemistry 2004;43:11998–2008
- Backman TW, Cao Y, Girke T. ChemMine tools: an online service for analyzing and clustering small molecules. Nucleic Acids Res 2011;39:W486–91
- de Clercq E, Descamps J, de Somer P, Holyacute A. (S)-9-(2,3-Dihydroxypropyl)adenine: an aliphatic nucleoside analog with broad-spectrum antiviral activity. Science 1978;200:563–5
- Wood MW, Rosqvist R, Mullan PB, et al. SopE, a secreted protein of Salmonella dublin, is translocated into the target eukaryotic cell via a sip-dependent mechanism and promotes bacterial entry. Mol Microbiol 1996;22:327–38
- Hardt WD, Urlaub H, Galan JE. A substrate of the centisome 63 type III protein secretion system of Salmonella typhimurium is encoded by a cryptic bacteriophage. P Natl Acad Sci USA 1998;95:2574–9
- Bakshi CS, Singh VP, Wood MW, et al. Identification of SopE2, a Salmonella secreted protein which is highly homologous to SopE and involved in bacterial invasion of epithelial cells. J Bacteriol 2000;182:2341–4
- Stender S, Friebel A, Linder S, et al. Identification of SopE2 from Salmonella typhimurium, a conserved guanine nucleotide exchange factor for Cdc42 of the host cell. Mol Microbiol 2000;36:1206–21
- Stevens MP, Friebel A, Taylor LA, et al. A Burkholderia pseudomallei type III secreted protein, BopE, facilitates bacterial invasion of epithelial cells and exhibits guanine nucleotide exchange factor activity. J Bacteriol 2003;185:4992–6