
Abstract
Context: Triosephosphate isomerase (TIM) is a ubiquitous enzyme that has been targeted for the discovery of new small molecular weight compounds used against Trypanosoma cruzi, the causative agent of Chagas disease. We have identified phenazine and 1,2,6-thiadiazine chemotypes as novel inhibitors of TIM from T. cruzi (TcTIM).
Objective: Study the mechanism of TcTIM inhibition by a phenazine derivative and by a 1,2,6-thiadiazine derivative.
Methods: We performed biochemical and theoretical molecular docking studies to characterize the interaction of the derivatives with wild-type and mutant TcTIM.
Results and conclusion: At low micromolar concentrations, the compounds induce highly selective irreversible inactivation of parasitic TIM. The molecular docking simulations indicate that the phenazine derivative likely interferes with the association of the two monomers of the dimeric enzyme by locating at the dimer interface, while 1,2,6-thiadiazine could act as an inhibitor binding to a region surrounding Cys-118.
Introduction
Trypanosoma cruzi (T. cruzi) has been identified as the causative agent of Chagas disease, an endemic disease widely disseminated in the American continent, which affects 9.8–11.0 million people, leaving 60 million people at riskCitation1. The therapies currently available have shown to be inadequate with respect to safety, efficacy, resistance, toxicity, difficulty of administration (in impoverished conditions) and cost issues. Despite the urgent need for new drugs, most pharmaceutical companies have neglected this disease, largely because of market policies.
The dependence of T. cruzi on glycolysis makes the enzymes participating in this pathway as excellent drug targets. Particularly, triosephosphate isomerase from Trypanosoma cruzi (TcTIM) has been widely investigated searching small molecular weight compounds that could act as selective inhibitors, thus, killing the parasiteCitation2–4.
TIM catalyzes the isomerization of glyceraldehyde-3-phosphate and dihydroxyacetone phosphate in the fifth step of the glycolytic pathway. Most of the described TIMs are homodimers and each monomer consists of eight parallel β-strands, surrounded by eight α-helices forming the classical α/β-barrel structure. In TcTIMCitation5, the interface between monomers represents a significant portion of the molecular surface area of each polypeptide chain: approximately 1496 Å2 in TcTIMCitation5. In all organisms, only the dimeric form of the enzyme is catalytically activeCitation6,Citation7.
We examined the interface of TIM from three different trypanosomatids, T. cruzi, Trypanosoma brucei (TbTIM) and Leishmania mexicana (LmTIM), and five other species, yeast, chicken, Homo sapiens (HsTIM), Plasmodium falciparum and Entamoeba histolytica. Our data show that the structure of the interface is highly conserved in the enzymes from the trypanosomatids (TcTIM, LmTIM and TbTIM), and differs significantly from that of the other species analyzedCitation8. In fact, the identity of the 32 interfacial residues of TcTIM, TbTIM and LmTIM with the homologous HsTIM is approximately 52%. TIMs from T. cruzi, T. brucei and L. mexicana have 73.9% sequence identity and 92.4% similarityCitation9. The potential use of protein–protein interfaces of homodimeric enzymes, as targets for drug discovery, has been reviewedCitation10.
As part of our ongoing program in the search of molecules that could provide leads in the design of a new drug for the treatment of Chagas diseaseCitation8,Citation9,Citation11,Citation12, we undertook a large screening for inhibitors of TcTIM, using nearly 230 compounds from an in-house chemical libraryCitation13. We identified four compounds (1–4, ), belonging to the 1,2,4-thiadiazole, phenazine and 1,2,6-thiadiazine chemotypes, that displayed selectivity for TcTIM over HsTIM. Moreover, these compounds are active on the whole parasiteCitation13. Recent studies of the mechanism of inactivation of compound 1 using a chimeral and a mutant TcTIMCitation14 have demonstrated that it is a tight-binding inhibitor.
Figure 1. Chemical structures of 1,2,4-thiadiazole (1), phenazine (2) and 1,2,6-thiadiazines (3, 4)Citation13.
In this work, we characterize the mechanism by which compounds 2 and 3 cause inhibition of TcTIM. Our data show that they are non-competitive inhibitors that induce the dissociation of two monomers, albeit they do not inhibit their association after they are unfolded by high guanidine chloride concentrations. In order to gain insight into which residues of the enzyme participate in the action of compounds 2 and 3, we studied their effects on a chimeric TIM.
We also examined the combined action of compounds 1, and 2 or 3 on TcTIM. We further show, using in silico molecular docking studies that compounds 1 and 3 bind to TcTIM at a site distinct from the substrate binding site, i.e. the region surrounding Cys-118, while compound 2 binds to TcTIM in a hydrophobic pocket localized at the interface between monomers.
Methods
Expression and purification of proteins
TcTIMCitation15, TbTIMCitation16, LmTIMCitation17 and TcTIM 2,3,5-8 (a chimeral protein that contains amino acids 1 to 35 and 92 to 119 of TbTIM and all the rest from TcTIMCitation18), were expressed in Escherichia coli and purified as described in the referred literature. After purification, the enzymes, dissolved in 100 mM triethanolamine, 10 mM EDTA and 1 mM dithiothreitol (pH 8), were precipitated with ammonium sulfate (75% saturation) and stored at 4 °C. Before use, extensive dialysis against 100 mM triethanolamine/10 mM EDTA (pH 7.4) was performed. Protein concentration was determined by absorbance at 280 nm. The ε (M−1 cm−1) were 36 440 for TcTIM and TcTIM 2,3,5-8, and 33 460 for TbTIM and LmTIMCitation19. Alternatively, protein concentration was measured using Bradford assay (Fermentas®, Vilnius, Lithuania) following manufacturer’s recommendations.
Activity assays
Activity was determined following the conversion of glyceraldehyde 3-phosphate into dihydroxyacetone phosphateCitation2. The decrease in absorbance at 340 nm was followed in a multicell Hewlett-Packard spectrophotometer at 25 °C. The reaction mixture (1 mL, pH 7.4) contained 100 mM triethanolamine, 10 mM EDTA, 0.2 mM NADH, 1 mM glyceraldehyde 3-phosphate (GAP) and 0.9 units of α-glycerol phosphate dehydrogenase (α-GPDH). The reaction was initiated by addition of 5 ng/mL of the corresponding TIM or the corresponding TIM pre-incubated with the studied compounds. In these cases, inhibition assays, the enzymes were pre-incubated for 2 h at 37 °C with the studied compounds. To calculate the kinetic parameters, we used increasing GAP concentrations from 0.05 to 10 mM. The data were adjusted according to the Michaelis–Menten model and the values of Km and Vmax were calculated by non-linear regression.
TcTIM dimer disruption evaluation
Formaldehyde-crosslinking-SDS-PAGE electrophoresis assays were performed to check the dimeric assembly of TcTIM. Protein (5 µg/20 µL) in 100 mM triethanolamine/10 mM EDTA (pH 7.4) was incubated for 2 h at 37 °C with and without the studied compounds. The reaction mixture was then exposed to formaldehyde 1% for 20 min. The samples were quenched with 5 µL of 4× sample buffer (Tris/HCl 0.25 M, SDS 8%, glycerol 40%, β-mercaptoethanol 20%, bromophenol blue 0.1%) and were heated at 100 °C for 5 min. The samples were analyzed by SDS-PAGE using a 14% acrylamide gel. A Page Ruler Prestained Protein Ladder (Fermentas®, Vilnius, Lithuania) was included as a standard to determine the molecular weight of each protein conformation. Proteins were visualized by staining with Coomassie brilliant blue (Sigma-Aldrich Corp., St. Louis, MO).
Effect of compounds 2 and 3 on the stability of TcTIM dimers
The enzyme was incubated at concentrations between 6 × 10−4 and 6 × 10−1 µM for 2 h at 37 °C with and without of compounds 2 or 3 (26 and 13 μM, respectively), and at that time the remaining activity was determined.
Folding/reactivation assay
TIM (TcTIM, HsTIM or TbTIM, 0.5 mg/mL) was denatured with guanidine chloride (6 M) in a final volume of 200 μL for 1 h. A portion (10 μL) of this solution was diluted to a final volume of 1 mL containing increasing concentrations of compounds 2 or 3. After 15 and 60 min, 1 μL of mixture was transferred to 1 mL of the activity assay mixture (see the Section “Activity assays”).
Competition assay
In this case, the enzyme was pre-incubated simultaneously with two inhibitors at various concentrations. On the one hand, compound 2 was assayed in combination with inhibitor 1 and, on the other hand, the same experiment was conducted for compound 3 combined with compound 1. The test was performed under the same conditions as the classic activity assay. The reaction was initiated by addition of 5 ng/mL of TcTIM or TcTIM pre-incubated with the mixtures of the studied compounds. In these last cases the enzyme was pre-incubated for 2 h at 37 °C with the mixture of the studied compounds. The assayed doses for the mixtures of compounds were: (i) (IC50/2) of compound 1 + (IC50/2) of compound 2 or 3; (ii) IC50 of compound 1 + (IC50/2) of compound 2 or 3; (iii) IC50 of compound 1 + IC50 of compound 2 or 3; (iv) 1.5 × IC50 of compound 1 + IC50 of compound 2 or 3; (v) 1.5 × IC50 of compound 1 + 1.5 × IC50 of compound 2 or 3; (vi) 2 × IC50 of compound 1 + 2 × IC50 of compound 2 or 3. From the activity data the inhibition curve for the mixture of compounds was plotted, as sum of units of IC50 versus percentage enzymatic inhibition, and compared with the curve of compound 1.
Theoretical calculations
The molecular structures of compounds 1–3 were fully optimized in vacuo at the DFT level using the B3LYP functional with the 6-31 G(d,p) basis set. Molecular docking of compounds 1–3 into the three-dimensional X-ray structure of TcTIM (PDB ID: 1TCD) was carried out with Autodock 4.2 (La Jolla, CA) using the implemented empirical free energy function and the Lamarckian Genetic Algorithm. The AutoDockTools package (La Jolla, CA) was employed to generate the docking input files and to analyze the docking resultsCitation20. Gasteiger-Marsilli charges were used for the protein and ligands. Because the location of the compounds in the enzyme was unknown, a grid map with 126 × 126 × 126 points and a grid-point spacing of 0.65 Å was applied initially in order to explore the entire protein surface. The maps were centered on the macromolecule. Once the binding site was determined, the calculations were refined using a grid box size of 60 × 60 × 66 points with a spacing of 0.375 Å. In these cases, the maps were centered in the ligand-binding site that had been previously determined. Each docking consisted of 50 independent runs, with an initial population of 150 individuals, a maximum number of 2.5 × 105 energy evaluations and a maximum number of 27 000 generations. Mutation and crossover were applied to the population at rates of 0.02 and 0.80, respectively. For the remaining parameters, default values were used. Results differing by less than 2.0 Å in root-square-deviation were grouped into the same cluster.
Results
The action of compounds 2 and 3 on different TIMs
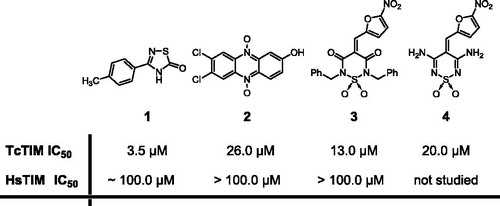
We studied the effect of different concentrations of compounds 2 and 3 on the activity of TcTIM. Our results show that, although both compounds are effective inhibitors of TcTIM (), compound 3 (with an IC50 of 13 µM) is about two times more effective than compound 2. The effect of different concentrations of compounds 2–4 on the activity of TcTIM is shown in . It is important to note that the activity of HsTIM is not affected by 100 µM of compounds 2 or 3, and that the IC50 of compound 1 on the activity of HsTIM is around 100 µM (). To further examine the selectivity of these compounds for TcTIM, we assessed their effect on the activity of LmTIM and TbTIM. The results show that phenazine 2, at a concentration of 100 µM, induced a rather small inhibition of the latter enzymes (), and that 1,2,6-thiadiazine 3 induced a higher level of inhibition, at a concentration of 100 µM, at least against the enzyme from T. brucei (). Thus, compound 2 seems to be highly selective for TcTIM. The use of higher concentrations of compound 3 was limited by its solubility in the milieu assay consequently the percentage of TcTIM inhibition by this compound never rises above ca. 80%.
Figure 2. (a) Dose–response curve of the inhibition of TcTIM activity by compounds 2, 3 and 4. (b) Dose–response curve for the inhibition by compound 3 of TcTIM, TbTIM and TcTIM 2,3,5-8 (chimera).
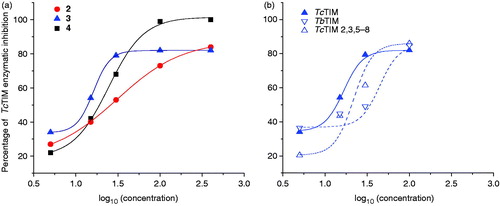
Table 1. Classic inhibition assay of Tc, Hs, Lm and Tb TIMs to evaluate the selectivity of the inhibitor compounds.
The kinetic studies of TcTIM with and without compounds 2 and 3, using different concentrations of glyceraldehyde 3-phosphate, indicated that these compounds are non-competitive inhibitors of the enzyme.
Some of the most powerful inhibitors of TcTIM act by lowering the association between the two monomers of the enzymeCitation21. Accordingly, we explored if compounds 2 and 3 exert their actions through the perturbation of monomer–monomer association. Different concentrations of TcTIM were incubated with the compounds for 2 h and the activity was measured (). The data shown in suggest that compounds 2 and 3 exert their actions by favoring the dissociation over the association reaction of TIM monomers; the presence of compound 2 increased the concentration of TcTIM by 10-fold at which equivalent activities were observed; and, for compound 3, a four-fold increase was required.
Figure 3. Effects of compounds 2 and 3 on the stability of TcTIM. (a) The enzyme was incubated at the indicated concentrations for 2 h at 37 °C with (▪) and without (•) 26 µM of compound 2 and the activity of each of the samples was measured. (b) The enzyme was incubated at the indicated concentrations for 2 h at 37 °C with (▪) and without (•) 13 µM of compound 3 and the activity of each samples was measured.
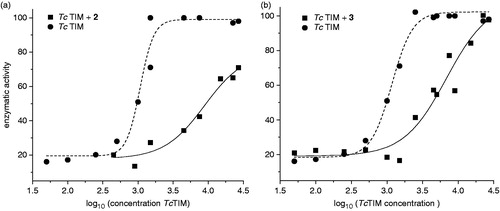
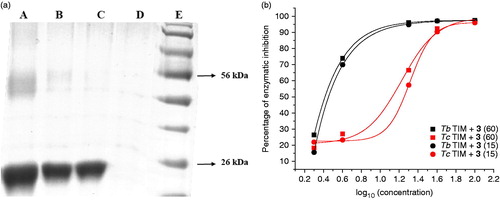
Cross-linking experiments of TcTIM followed by SDS-PAGE experiments yielded a band of 54 kDa that corresponds to the dimer formation. A dose dependent disappearance of this band was observed using increasing amounts of the compounds. These experiments suggest that the compounds act as disruptors of the dimeric form of TcTIM (see example in ). To examine if the disruption of the dimeric form of TIMs is due to the association of compounds 2 or 3 with its monomers, we denatured the TIMs in 6 M guanidine chloride (GdnHCl). In a second step of the experiment, the denaturing mixture was diluted in buffer containing different concentrations of the studied compounds. As previously shown by Zomosa-Signoret et al.Citation22, the removal of the denaturant, GdnHCl, by dilution leads to the refolding and association of the monomers to form the catalytically active dimer. This reaction can be followed, by measuring the appearance of activity through time.
Figure 4. (a) Electrophoresis analysis (14 % SDS-PAGE) of formaldehyde-crosslinking of TcTIM in the presence of compound 1 (see “Methods” section for experimental conditions). Lane A: TcTIM 5 µg/20 µL; Lane B: TcTIM 5 µg/20 µL incubated with 3.5 µM of compound 1; Lane C: TcTIM 5 µg/20 µL incubated with 35.0 µM of compound 1; Lane D: compound 1 at 35.0 µM; Lane E: page ruler prestained protein ladder, Fermentas® SM0671. (b) Effect of compound 3 on the formation of active TcTIM and TbTIM from GdnHCl unfolded monomers. The experiment was performed as described in the “Methods” section. Activity was measured 15 (•) and 60 (▪) min after the denaturing mixture was diluted 100 - fold.
Compound 2 did not inhibit the formation of active HsTIM or TbTIM from their corresponding unfolded monomers at the assayed doses, while with TcTIM, phenazine 2 inhibited the dimer formation from the unfolded monomers with the same IC50 as the wild enzyme ().
On the other hand, 1,2,6-thiadiazine 3 showed a different behavior. It did not inhibit the formation of active HsTIM from the corresponding unfolded monomers at the assayed doses, while TcTIM dimer formation was inhibited with an IC50 similar to that observed with the wild enzyme: 17 ± 1 μM versus 13 ± 1 μM (, ), respectively. It is also noteworthy that compound 3 effectively inhibited the formation of active TbTIM dimers from its corresponding unfolded monomers with an IC50 for the inhibition of dimer formation of 0.7 μM and an IC90 of 8.0 μM (). Different inhibition curves for TcTIM and TbTIM could be associated to different inhibition mechanisms.
Attempts to identify the amino acid regions of TcTIM that make it susceptible to the action of compounds 2 and 3
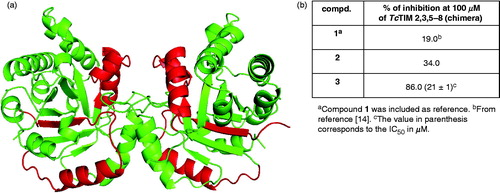
In order to identify the regions of TcTIM that make it susceptible to the action of compounds 2 and 3, two different approaches were performed. Recently, we reportedCitation14,Citation18 that it is possible to use chimeras of TcTIM and TbTIM in order to identify the amino acids or amino acid sequences that are involved in a given property of the enzymes. Thus, we examined the effect of compounds 2 and 3 on a TcTIM in which regions 1 (residues 1–35) and 4 (residues 92–119) were replaced by the corresponding regions of TbTIM, namely TcTIM 2,3,5-8 (). This chimera was less sensitive to phenazine 2 than TcTIM (). However, 1,2,6-thiadiazine 3 was a good inhibitor of this chimera, displaying an IC50 of 21 μM, which is an intermediate value between those corresponding to wild type TcTIM and TbTIM ( and ).
Figure 5. (a) Graphical representation of TcTIM showing the regions of TbTIM (highlighted) that were grafted onto TcTIM to generate the chimera TcTIM 2,3,5-8. (b) Classic inhibition assay, using modified TcTIM, to evaluate the selectivity of the inhibitors 2 and 3.
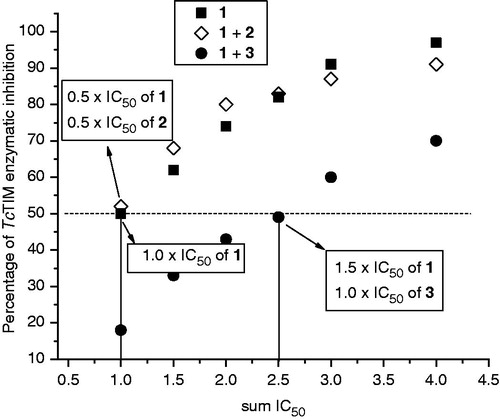
In addition, we performed inhibition assays using compounds 1 and 2, or 1 and 3, simultaneously. It was observed () that compounds 1 and 2 had additive effects since the 1 + 2 curve superimposed on the compound 1 curve. However, compounds 1 and 3 had competitive effects, given that inhibitor 1 required 1.5-fold higher doses to inhibit 50% of TcTIM, when it was incubated in presence of the IC50 concentration of compound 3. These results could indicate that compounds 1 and 2 are positioned in different locations on TcTIM, and that compounds 1 and 3 are situated in nearby sites on the enzyme.
Figure 6. TcTIM inhibition assays using mixtures of compounds 1 + 2(◊) and 1 + 3 (•). In each run we used concentrations which were multiples or submultiples of the IC50 of the compound 1 together with variable amount of compounds 2 or 3, which, correspondingly, were multiples or submultiples of the IC50. The values were plotted as sum of the IC50 used (sum IC50). The behavior of compound 1 alone (▪)Citation14 was included as reference.
Theoretical calculations
In order to investigate the binding modes and inhibitory mechanism of compounds 1–3 for TcTIM, molecular docking studies were performed. The predicted binding modes of the three species with TcTIM are illustrated in and the detailed information about the interactions between the ligands and the protein is illustrated in .
Figure 7. (a–c) Predicted binding mode of compounds 1–3 according to the data obtained from the molecular docking studies; the contact residues around the binding site are shown, the circles indicate the catalytic sites. (d–f) The contact residues, which are defined as those within 3.0 Å of each ligand, are shown and labeled by the type and number. The TcTIM dimer is represented by the solvent-accessible-surface area (SASA) model. Compounds are represented as VDW models in a–c and as stick models in d–f.
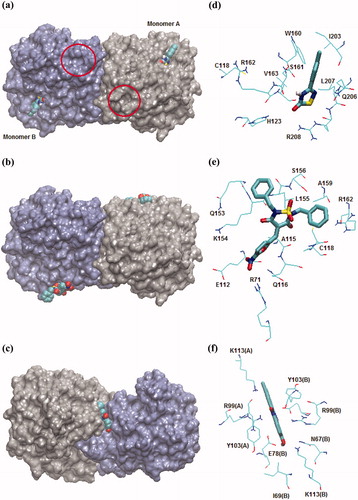
As shown in , compounds 1 and 3 bind to both monomers of TcTIM. According to these compounds bind to adjacent regions surrounding Cys-118. Compound 1 makes hydrophobic interactions with Ile-203, Leu-207 and Val-163. The tolyl-substituent of the ligand establishes π–π stacking interactions with the indole group of Trp-160. It also has hydrophilic interactions with His-123, Arg-162, Arg-208, Ser-161 and Gln-206. Among these residues, the lateral chain nitrogen atoms NH and NH2 of Arg-208 form hydrogen bonds with the heterocyclic sulfur atom and the oxygen atom of the carbonyl group of the ligand. Additionally, the backbone oxygen atom of Ser-161 forms a hydrogen bond with the NH atom of 1 (). As depicted in , compound 3 makes hydrophobic interactions with Leu-155, Ala-159 and Ala-115. The nitrogen atoms, Nε2 of Gln-116 and NH2 of Arg-71, establish hydrogen bonds with the oxygen atom and the nitro group of the furane ring of the ligand, respectively. Moreover, the OH group of Ser-156 forms a hydrogen bond with an oxygen atom of the sulfonyl moiety of 3. The binding of these compounds, at a site other than the active site (marked with red circles in ), is consistent with the experimental results which show that compounds 1 and 3 are non-competitive inhibitors of TcTIM. In addition, regions near Cys-118 have been described as sites that, upon occupation, are able to alter the dimer interface [9]. Therefore, the mechanism of inhibition of these compounds could be mediated by disruption of the hydrogen bond network between one monomer and a flexible loop (loop 3) of the other monomer that is essential for dimer integrity. Interestingly, the residues that form the binding pocket of compounds 1 and 3 show a sequence identity of 51% between TcTIM and HsTIM, and thus, are potential sites for the design of selective compounds against the parasitic enzyme. According to the docking results, the antagonistic effect caused by 3 in the activity of 1 in the co-incubation assay could be explained considering the binding site of these compounds. Since both molecules bind to adjacent regions around Cys-118, they could compete for access to the binding site. The docking results of 3 are also consistent with the experimental data on the inhibition of the chimeral TIM by this compound. Because the binding site of 1,2,6-thiadiazine 3 does not involve the amino acids replaced in the chimeral construct, a differential sensitivity of chimeral TIM for this derivative would not be expected.
On the other hand, compound 2 binds to TcTIM in a deep trench localized at the interface between monomers (). Accordingly, this compound could act as TcTIM inhibitor by causing steric hindrance around the monomer interface perturbing the interaction and association between monomers, as was determined in the experimental assays. shows that compound 2 is stabilized in its binding pocket by hydrophobic and electrostatic interactions. The phenol ring of Tyr-103 in monomers A and B forms π–π stacking interactions with the phenazine ring of the ligand. The nitrogen atoms NH2 of Arg-99, in both monomers, form hydrogen bonds with the oxygen atom of each N-oxide moiety of 2. Moreover, the OH moiety of the inhibitor establishes hydrogen bonds with the Nζ atom of Lys-113 and the Oε1 atom of Glu-78 of monomer B. The positively charged NH3 group of Lys-113 in monomer A is properly located to establish electrostatic interactions with the negative charge density around the chloride substituents of the phenazine ring. The interaction of compound 2 with Glu-78 at the interface of TcTIM is an interesting finding since this residue is replaced by Ile in HsTIM. Therefore, designing molecules with groups bearing a positive charge which are able to interact specifically with negatively charged residues would be a good strategy for obtaining selective and better inhibitors of TcTIM. The results we obtained with compound 2 are consistent with the experimental finding that the chimeral TIM is less sensitive to this compound. In fact, the binding site of 2 spans amino acids 67 to 113 of each monomer including most of the residues used in the chimera construction, i.e. amino acids 92–119. In addition, the binding of 2 to a different site than that of compound 1 explains the observation that the activity of 1 was not affected by the addition of 2 in the experiments performed to analyze the combined effect of these two inhibitors.
Discussion
Using a large screening assay that monitors the inactivation of TcTIM, we identified the phenazine derivative 2 and the 1,2,6-thiadiazine derivatives 3 and 4 as small molecules with potential anti-T. cruzi activityCitation13. We found that compound 2 selectively inhibits the T. cruzi enzyme, without affecting the TIMs from H. sapiens and Leishmania sp. and with rather meager effects on TIM from T. brucei. On the other hand, compound 3 inhibits TcTIM as well as TIM from T. brucei, without any effect on the H. sapiens enzyme and with rather scarce effects on Leishmania sp. enzyme. The crosslinking experiments followed by SDS-PAGE verified the TcTIM dimer disruption by the studied compounds. As shown here, compounds 2 and 3 diminish the stability of the TcTIM-dimer and prevent its formation from unfolded monomers. Additionally, at submicromolar concentrations, only compound 3 inhibits the formation of TbTIM, but not that of the HsTIM, from their respective unfolded monomers, suggesting that the two trypanosomal TIMs have a binding site for compound 3. The data with the chimera gives insight into which residues could contribute to the binding of the compounds to TcTIM. The docking models are consistent with the experimental data providing us with interesting molecular-level information, not only for understanding the mechanism of these novel inhibitors, but also for future prospects in rational drug design.
Declarations of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper. The authors thank the financial supports from Cooperación Mexicana para el Desarrollo, from the Dirección General de Asuntos del Personal Académico, U.N.A.M. No. IN221812-3 (R. P-M.), from the Consejo Nacional de Ciencia y Tecnología, México No. 167823 (R.P-M.), and from RIDIMEDCHAG-CYTED. The authors also thank ANII for the scholarships to GA and JM.
Related Research Data
References
- Schofield CJ, Jannin J, Salvatella R. The future of Chagas disease control. Trends Parasitol 2006;22:583–8
- Gomez-Puyou A, Saavedra-Lira E, Becker I, et al. Using evolutionary changes to achieve species-specific inhibition of enzyme action – studies with triosephosphate isomerase. Chem Biol 1995;2:847–55
- Velanker SS, Ray SS, Gokhale RS, et al. Triosephosphate isomerase from Plasmodium falciparum: the crystal structure provides insights into antimalarial drug design. Structure 1997;5:751–61
- Cortés-Figueroa AA, Pérez-Torres A, Salaiza N, et al. A monoclonal antibody that inhibits Trypanosoma cruzi growth in vitro and its reaction with intracellular triosephosphate isomerase. Parasitol Res 2008;102:635–43
- Maldonado E, Soriano-García M, Moreno A, et al. Differences in the intersubunit contacts in triosephosphate isomerase from two closely related pathogenic trypanosomes. J Mol Biol 1998;283:193–203
- Waley SG. Refolding of triose phosphate isomerase. Biochem J 1973;135:165–72
- Zabori S, Rudolph R, Jaenicke R. Folding and association of triose phosphate isomerase from rabbit muscle. Z Naturforsch C 1980;35:999–1004
- Olivares-Illana V, Pérez-Montfort R, López-Calahorra F, et al. Structural differences in triosephosphate isomerase from different species and discovery of a multitrypanosomatid inhibitor. Biochem 2006;45:2556–60
- Olivares-Illana V, Rodríguez-Romero A, Becker I, et al. Perturbation of the dimer interface of triosephosphate isomerase and its effect on Trypanosoma cruzi. PLoS Neglected Trop Dis 2007;1:e01
- Cardinale D, Salo-Ahen OM, Ferrari S, et al. Homodimeric enzymes as drug targets. Curr Med Chem 2010;17:826–46
- Cerecetto H, González M. Anti-T. cruzi agents: our experience in the evaluation of more than five hundred compounds. Mini Rev Med Chem 2008;8:1355–83
- Benitez D, Cabrera M, Hernández P, et al. 3-Trifluoromethylquinoxaline N,N'-dioxides as anti-trypanosomatid agents. Identification of optimal anti-T. cruzi agents and mechanism of action studies. J Med Chem 2011;54:3624–36
- Alvarez G, Aguirre-López B, Varela J, et al. Massive screening yields novel and selective Trypanosoma cruzi triosephosphate isomerase dimer-interface-irreversible inhibitors with anti-trypanosomal activity. Eur J Med Chem 2010;45:5767–72
- Alvarez G, Aguirre-López B, Cabrera N, et al. 1,2,4-thiadiazol-5(4H)-ones: a new class of selective inhibitors of Trypanosoma cruzi triosephosphate isomerase. Study of the mechanism of inhibition. J Enzyme Inhib Med Chem 2012preprint. doi: 10.3109/14756366.2012.700928 (Early Online: http://informahealthcare.com/toc/enz/0/0)
- Ostoa-Saloma P, Garza-Ramos G, Ramírez J, et al. Cloning, expression and purification of triosephosphate isomerase from Trypanosoma cruzi. Eur J Biochem 1997;244:700–5
- Borchert TV, Pratt K, Zeelen JP, et al. Overexpression of trypanosomal triosephosphate isomerase in Escherichia coli and characterization of a dimer interface mutant. Eur J Biochem 1993;211:703–10
- Kohl L, Callens M, Wierenga RK, et al. Triosephosphate isomerase of Leishmania mexicana mexicana. Cloning and characterization of the gene, overexpression in Escherichia coli and analysis of the protein. Eur J Biochem 1994;220:331–8
- García-Torres I, Cabrera N, Torres-Larios A, et al. Identification of amino acids that account for long-range interactions in two triosephosphate isomerases from pathogenic trypanosomes. PLoS ONE 2011;6:e18791
- Pace CN, Vajdos F, Fee L, et al. How to measure and predict the molar absorption coefficient of a protein. Protein Sci 1995;4:2411–23
- Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91
- Téllez-Valencia A, Avila-Ríos S, Pérez-Montfort R, et al. Highly specific inactivation of triosephosphate isomerase from Trypanosoma cruzi. Biochem Biophys Res Commun 2002;295:958–63
- Zomosa-Signoret V, Hernández-Alcántara G, Reyes-Vivas H, et al. Control of the reactivation kinetics of homodimeric triosephosphate isomerase from unfolded monomers. Biochemistry 2003;42:3311–18