
Abstract
A series of eight thiosemicarbazide derivatives was examined for cytotoxicity in breast cancer cell cultures. Among them, 4-benzoylthiosemicarbazides proved to be only slightly less potent than chlorambucil in both MDA-MB-231 and MCF-7 lines. In contrast, 4-aryl/alkylthiosemicarbazides revealed significantly lower cytotoxicity effect. Subsequently, all titled compounds were tested as potential human topoisomerase I and II (topo I and topo II) inhibitors. Mechanistic studies revealed that tested thiosemicarbazides act as both topoisomerase I and topoisomerase II inhibitors. Among them, the best inhibitory activity was found for 4-benzoylthiosemicarbazides (1 and 2) with IC50 at 50 µM against topo II.
Introduction
Thiosemicarbazides, a large group of thiourea derivatives, exhibits various biological activities and have therefore attracted considerable pharmaceutical interestCitation1,Citation2. They comprise an intriguing class of chelating molecules which possess a wide range of beneficial medicinal properties and exhibit versatile structural features. They have been evaluated as antiviral, antibacterial and anticancer therapeutics over the last 50 yearsCitation1,Citation2. Although a number of reports are available on biological activity of thiosemicarbazides, study on topoisomerase activity is still scarcely explored. The chemotherapeutic potential of DNA topoisomerase inhibitors has been clearly established in recent yearsCitation3–5. The antitumor properties of several clinically used drugs (e.g. amsacrine, doxorubicin, actinomycin D, mitoxantrone, topotecan, irinotecan) are believed to result from their ability to both bind to DNA and to interfere with the catalytic activities of topoisomerasesCitation3–5. Therefore, designing drugs able to inhibit topoisomerase activities is considered a worthwhile goal in the search for new antitumor agents. DNA topoisomerases fall into two general categories (types I and II) based on biochemical mechanism responsible for DNA strand passageCitation6. Type I enzymes catalyze single-stranded DNA cleavage and strand passage in the absence of ATP, while type II topoisomerases mediate double-stranded breakage with passage of another helix through the transient break by a process requiring energy from the hydrolysis of ATP. The DNA strand passage reactions mediated by topoisomerases are essential for maintaining the appropriate state of DNA supercoiling in the cell, for efficient replication of DNA, and for the separation of daughter DNA molecules during cell divisionCitation6,Citation7. Type I and II topoisomerases are found in all cells and act in concert to maintain the optimal DNA conformation in the cell. Small molecule inhibitors targeting both types of topoisomerase are an important component in the chemotherapeutic arsenal used to treat cancerCitation3–5,Citation8. Unfortunately, due to serious side effects and/or development of resistant cancer cells, new improved versions are needed. Besides, most of these inhibitors have large molecular weight and complex structure, which make it difficult and expensive to synthesize these compounds at an industrial scale. Therefore, it is necessary to search for new types of topoisomerase inhibitors that can be easily synthesized with better therapeutic index.
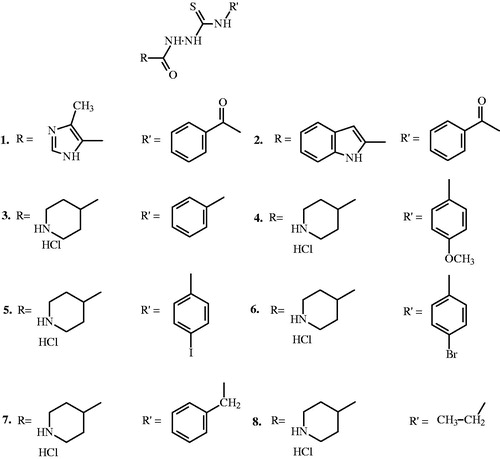
Very recently, promising results for thiosemicarbazone derivatives, closely related compounds for thiosemicarbazides, as potential topoisomerase inhibitors were publishedCitation9–11. Inspired by these optimistic results, we have synthesized and investigated the antiproliferative activity of thiosemicarbazide derivatives (1–8) () in both MDA-MB-231 and MCF-7 breast cancer cells and assessed their ability to act as inhibitors of topoisomerases I and II (topo I and II).
Figure 1. Structure of compounds 1–8.
Methods
Reagents and materials
4-Aryl/alkyl-1-(piperidin-4-yl)-carbonylthiosemicarbazides and 4-benzoyltiosemicarbazides used in this study (1–8) were synthesized as described in our previous papersCitation12,Citation13. Calf thymus DNA, ethidium bromide (EtBr), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), genistein, netropsin were purchased from Sigma (St. Louis, MO). Topoisomerase I and topoisomerase II, supercoiled pHOT1 DNA, supercoiled pRYG DNA, etoposide, camptothecin were purchased from TopoGEN (Port Orange, FL). Stock cultures of breast cancer MCF-7 and MDA-MB-231 were purchased from the American Type Culture Collection (Rockville, MD). Dulbecco's minimal essential medium (DMEM) and foetal bovine serum (FBS) used in cell culture were the products of Gibco (Life Technologies, Grand Island, NY). Glutamine, penicillin and streptomycin were obtained from Quality Biologicals Inc. (Gaithersburg, MD). [3H]Thymidine (6.7 Ci/mmol) was the product of NEN (Waltham, MA).
Cell culture
Cultured normal human skin fibroblasts, MDA-MB-231 and MCF-7 human breast cancer cells maintained in DMEM supplemented with 10%FBS, 50 U/ml of penicillin, 50 μg/ml of streptomycin at 37 °C. Cells were cultured in Costar flasks and subconfluent cells were detached with 0.05% trypsin and 0.02% EDTA in calcium-free phosphate buffered saline, counted in hemocytometers and plated at 5 × 105 cells per well of 6-well plates (Nunc, Panfield, NY) in 2 ml of growth medium (DMEM without phenol red with 10% CPSR1). Cells reached about 80% of confluency at day 3 and in most cases such cells were used for the assays.
Cytotoxic assay
To examine the effect of studied drugs on cultured normal human skin fibroblasts, MCF-7 and MDA-MB-231 breast cancer cells proliferation, the cells were seeded in 24-well tissue culture dishes at 1 × 105 cells/well with 1 ml of growth medium. After 48 h (1.8 ± 0.1 × 105 cells/well) plates were incubated with varying concentrations of compounds 1–8, chlorambucil and 0.5 μCi of [3H]thymidine for 24 h at 37 °C. Cells were rinsed three times with PBS, solubilized with 1 ml of 0.1 M sodium hydroxide containing 1% SDS, scintillation fluid (9 ml) was added and radioactivity incorporation into DNA was measured in scintillation counter.
Cell viability assay
The assay was performed according to the method of Carmichael using 3-(4,5-di-methylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT)Citation14. Confluent cells, cultured for 24 h with various concentrations of the studied compounds in 6-well plates were washed three times with PBS and then incubated for 4 h in 1 ml of MTT solution (5 mg/ml of PBS) at 37 °C in 5% CO2 in an incubator. The medium was removed and 1 ml of 0.1 M HCl in absolute isopropanol was added to the attached cells. Absorbance of converted dye in living cells was measured at a wavelength of 570 nm. Cell viability of breast cancer cells cultured in the presence of ligands was calculated as a percent of control cells.
Relaxation assay of topoisomerase I
Supercoiled pHOT1 DNA (0.5 μg) was incubated with four units of human topoisomerase I in relaxation buffer (10 mM Tris-HCl (pH 7.8), 1 mM EDTA, 0.15 M NaCl, 0.1% BSA, 0.1 spermidine, 5% glycerol), in the presence of varying concentrations of the test compounds. Reactions were carried out at 37 °C for 1 h and then terminated by the addition of sodium dodecyl sulfate (SDS) to 0.25% and proteinase K to 50 μg/ml. The reaction mixture was subjected to electrophoresis through a 0.8% agarose gel containing 0.5 mg/ml ethidium bromide in TAE buffer (40 mM Tris-borate and 1 mM EDTA). The gels were stained with ethidium bromide and photographed under UV light. For the quantitative determination of topoisomerase concentration activity, photographic negatives were scanned and the area representing supercoiled DNA, migrating as a single band at the bottom of the gel was measured using UVI-KS4000i gel documentation and analysis system (SyngenBiotech, Sacramento, CA). The concentrations of the inhibitor that prevented 50% of the supercoiled DNA from being converted into relaxed DNA (IC50 values) were determined by averaging the data from at least five experiments.
Relaxation assay of topoisomerase II
Supercoiled pRYG DNA (0.5 μg) was incubated with four units of human topoisomerase II in the cleavage buffer (30 mM Tris-HCl (pH 7.8), 50 mM KCl, 10 mM MgCl2, 3 mM ATP, 15 mM mercaptoethanol), in the presence of varying concentrations of the test compounds. Reactions were carried out at 37 °C for 1 h and then terminated by the addition of 2 μl 10% SDS and 2 μl 50 µg/ml proteinase K. The reaction mixture was subjected to electrophoresis through a 0.8% agarose gel containing 0.5 mg/ml ethidium bromide in TBE buffer (90 mM Tris-borate and 2 mM EDTA). The gels were stained with ethidium bromide and photographed under UV light. For the quantitative determination of topoisomerase concentration activity, photographic negatives were scanned and the area representing supercoiled DNA, migrating as a single band at the bottom of the gel was measured using UVI-KS4000i gel documentation and analysis system (SyngenBiotech, Sacramento, CA). The concentrations of the inhibitor that prevented 50% of the supercoiled DNA from being converted into relaxed DNA (IC50 values) were determined by averaging the data from at least five experiments.
Statistical analysis
In all experiments, the mean values for six independent experiments ± standard deviations (SD) were calculated, unless otherwise indicated. The results were submitted to statistical analysis using Students t-test, accepting p < 0.05, as significant.
Computational details
Physicochemical parameters were calculated using HyperChem8.0.3Citation15. Extensive conformational searches were carried out using the molecular mechanics level with the OPLSCitation16,Citation17 force field. The obtained most stable structures were subsequently optimized to the closest local minimum at the semi-empirical level using RM1 parametrizationCitation18. Convergence criteria were set to 0.1 and 0.01 kcal mol−1 Å−1 for OPLS and RM1 calculations, respectively.
Results
Cell viability of breast cancer cells was measured by the method of Carmichael et al. using tetrazolium saltCitation14. All the synthesized thiosemicarbazide compounds (1–8) and the positive control chlorambucil were evaluated for their ability to antiproliferative activities against breast cancer cells. Although growth inhibition was concentration-dependent in either cell line, it was more pronounced at shorter times, in MDA-MB-231 than MCF-7 (). The values of IC50 were relatively higher for 1 and 2 which possess a 4-benzoyl function. n terms of reduction in cell viability, the compounds 1–8 rank in both MCF-7 and MDA-MB-231 cells in the order 1 > 2 > 5 > 4 > 6 > 3 > 7 > 8 (). Among the derivatives, 4-benzoylthiosemicarbazides (1 and 2) in both MDA-MB-231 and MCF-7 proved to be only slightly less potent than chlorambucil, with IC50 values in MCF-7 of 124 ± 2 μM and 148 ± 2 μM, and in MDA-MB-231 of 120 ± 2 μM and 152 ± 2 μM, respectively, compared to 97 ± 2 μM for chlorambucil in MCF-7 and 92 ± 2 μM in MDA-MB-231 cells. In contrast, 4-aryl/alkyl-1-(piperidin-4-yl)-carbonylthiosemicarbazides (3–8) reveal significantly lower cytotoxicity against MDA-MB-231 and MCF-7 breast cancer cells with IC50 values of more than 300 μM ().
Table 1. IC50 values* (μM) of compounds 1–8 and chlorambucil in various cell lines using the MTT assay.
To analyze if the inhibition in cell viability was due to decreased cell proliferation, we measured DNA synthesis in presence of compounds 1–8 and chlorambucil. Measurement of [3H]thymidine incorporation during the DNA synthesis by proliferating breast cancer cells showed that these compounds inhibited DNA synthesis in a dose-dependent manner (). All of the tested compounds showed concentration-dependent activity, yet with different potency. Furthermore, the profiles of DNA synthesis obtained were similar between MCF-7 and MDA-MB-231 (). The concentrations of 1 and 2 needed to inhibit [3H]thymidine incorporation into DNA by 50% (IC50) in MDA-MB-231 were found to be 98 ± 2 μM and 124 ± 3 μM, respectively, suggesting lower cytotoxic potency compared to chlorambucil (IC50 = 49 ± 2 μM). The concentrations of 1, 2 and chlorambucil needed to 50% reduction in [3H]thymidine incorporation into DNA in breast cancer MCF-7 (IC50) were found to be 101 ± 2 μM, 127 ± 2 μM and 57 ± 2 μM, respectively. In contrast, 4-aryl/alkyl-1-(piperidin-4-yl)-carbonylthiosemicarbazides (3–8) are clearly much less active (). Because the antiproliferative effect of compounds 1 and 2 is independent of the estrogen receptor status of the breast cancer cells, these potent inhibitors are potential pharmacological agents for the treatment of both hormone responsive and nonresponsive breast cancer cells.
Figure 2. Cytotoxic effects of compounds 1–8 and chlorambucil on the cultured breast cancer MDA-MB-231 (A) and MCF-7 (B) cells as measured by inhibition of [3H]thymidine incorporation into DNA. Mean values ± S.D. of three independent experiments done in duplicates are presented.
![Figure 2. Cytotoxic effects of compounds 1–8 and chlorambucil on the cultured breast cancer MDA-MB-231 (A) and MCF-7 (B) cells as measured by inhibition of [3H]thymidine incorporation into DNA. Mean values ± S.D. of three independent experiments done in duplicates are presented.](/cms/asset/6d839635-fdb8-4b95-8a59-f02e6b16e202/ienz_a_768987_f0002_b.jpg)
The effect of the compounds 1–8 on the catalytic activities of purified human topo I and topo II was quantified by measuring the action on supercoiled plasmid DNA substrate as a function of increasing concentration of the ligands by the use of agarose gel electrophoresis. The DNA samples were treated with SDS and proteinase K to remove any covalently bound protein and were then resolved in a 1% agarose gel. The concentrations of the inhibitor that prevented 50% of the supercoiled DNA from being converted into relaxed DNA (IC50 values) were determined (). Camptothecin and etoposide, two well-known inhibitors of topoisomerases I and II, respectively, were used as positive references in these experiments. Enzymatic activity (relaxation of pRYG plasmid) of topo II was inhibited for 1 and 2 at a concentration of 50 μM (), a little decline compared to the positive control etoposide (IC50 = 30 μM). These results demonstrated also that 4-benzoylthiosemicarbazides (1–2) had topo I inhibitory activity at the concentration of 100 μM (positive control camptothecin IC50 = 20 μM). The compounds 3–6 were topoisomerase I and II inhibitors, with 50% inhibitory concentration IC50 = 250 μM (). Based on this result, it might be speculated that the phenyl ring fused into 1-(piperidin-4-ylcarbonyl)-thiosemicarbazide core is necessary to exert human topo I/II inhibition activity and in the aryl analogs series 3–6, inhibitory potency against topo I/II seems not related to the electronic effect (analog with an electron-donating 4-methoxy group (4) and analog with an electron-withdrawing 4-iodo group (5) had the same IC50 value). Human topo II inhibitory activity does not appear to be a sensitive function of the substituent on N-4 nitrogen atom of the thiosemicarbazide skeleton. As shown in , no inhibitory effect was observed for 7–8 even up to the concentration of 350 μM.
Table 2. Inhibitory activity of compounds 1–8, etoposide (ET) and camptothecin (CT) against human topoisomerases I and II.
To investigate the interaction of 4-benzoylthiosemicarbazides (1 and 2) with topoisomerase II or with substrate DNA, topoisomerase II or substrate DNA was increased in the reaction mixture and the inhibitory profiles were observed. When the amount of the enzyme was increased from 4 to 7 units, recovery of the enzyme activity was observed (results not shown). In contrast, increasing the amount of DNA showed no recovery of the enzyme activity. The results suggest that the drugs primarily interact with topoisomerase II and not with DNA. Consistent with this fact, inhibition of topoisomerase II catalytic activity by compounds 1 and 2 could be reversed by increasing the concentration of enzyme in the reaction mixture.
Discussion
The topoisomerase-targeting drugs can be classified as either topo poisons, which act by stabilizing enzyme-DNA cleavable complexes leading to DNA breaks, or topo catalytic inhibitors, which act at stages in the catalytic cycle of the enzyme where both DNA strands remain intact and no DNA strand breaks occurCitation3–5. Topoisomerase inhibitors can be divided into three categories based on specificity for the class of enzyme targeted (topoisomerase I, II or the more rare dual topo I/II inhibitors)Citation3–5. The compounds 1 and 2 do not promote DNA cleavage by topo I or II, in contrast to the reference classical poisons camptothecin (for topo I) and etoposide (for topo II). The intensity of the band corresponding to nicked DNA (for topo I) and linear DNA (for topo II) is not amplified in the presence of 4-benzoylthiosemicarbazides (1–2), implying that these compounds do not act as topo poisons. It is likely that the ability of 4-benzoylthiosemicarbazides (1–2) to inhibit the activity of topo I and topo II that we have observed () is simply due to blockade of the binding of these enzymes to DNA.
Many antitumor drugs including the bleomycins, the pluramycins and the aureolic acids (e.g. mithramycin, chromomycin and olivomycin) possess DNA-intercalating domainCitation19. The potential DNA-binding properties of compounds 1 and 2 have been assessed by measuring the displacement of ethidium bromide (EtBr) from calf thymus DNA. The 4-benzoylthiosemicarbazides (1 and 2) appeared totally inactive in the EtBr/DNA displacement assay at concentrations up to 100 μM (data not shown), contrasting with the effects of true intercalators such as doxorubicin or minor groove binders like netropsin and distamycin ACitation19,Citation20, which showed definite activities in assays at concentrations as low as 10 μM. The epipodophylloid-type compound etoposide, as well as a non-epipodophylloid type of topoisomerase II inhibitor, genistein, were found to be inactive in a displacement assay, as expected. Moreover, the known specific inhibitor of topoisomerase I, camptothecin, also proved negative in this EtBr displacement assay. These results indicate that 4-benzoylthiosemicarbazides (1–2) neither intercalate nor interact with the minor groove of DNA.
The degree to which compounds 1 and 2 inhibited cell growth in breast cancer cells was correlated to topoisomerase II-inhibiting activity. The data presented here favor a direct interaction between 4-benzoylthiosemicarbazides (1 and 2) and the topoisomerase II enzyme. Chemotherapeutic agents that target topoisomerases I and II can set in motion a series of biochemical changes that culminate in cell death, but only under certain conditionsCitation4,Citation5,Citation21. Whilst a range of signaling molecules have been implicated in cell death mediated by topoisomerase-interacting agents, generally their roles remain undefined. The realization that stabilization of topoisomerase–drug complexes was insufficient to ensure this ultimate fate has prompted major research in this area. One might speculate, therefore, that the abilities of 4-benzoylthiosemicarbazides (1 and 2) to compromise the overall catalytic activities of topoisomerase II impact on their cytotoxic potential via some as yet to be defined activation of an apoptotic program. Thus, in addition to inhibition of topoisomerase II, the cytotoxicity of 4-benzoylthiosemicarbazides (1 and 2) is associated with multiple mechanisms that may be responsible for inhibition of growth of tumor cells. These studies are underway in our laboratories and will be described in due course.
The above presented results of cytotoxicity assay () suggest that the position and the identity of individual substitutions are important parameters influencing the activity of studied thiosemicarbazides (1–8). In the structure of the thiosemicarbazide, the presence of benzoyl moiety at four position seemed to confer higher bioactivity than that of aryl or alkyl group. Following this finding, we have analyzed some structural and electronic parameters in detail that are frequently used in SAR analysis. However, the lack of statistically important relationship between the bioactivity of 4-benzoylthiosemicarbazides (1 and 2) and parameters such as surface area (SA), volume (V), refractivity (Rf), polarizabilitiy (α), moment dipole (µ), the highest occupied molecular orbital energy (EHOMO), the lowest unoccupied molecular orbital energy (ELUMO), the difference between HOMO and LUMO energy levels (HLG), hardness (η) obtained from the equation η = (ELUMO − EHOMO)/2, Mulliken electronegativity (χ) obtained from the equation χ =−(EHOMO + ELUMO)/2, total energy (ET), binding energy (EB), isolated atomic energy (EIA), electronic energy (EE), core–core interaction (IC--C) and heat of formation (HF) was observed. On the other hand, in analyzing the cytotoxic activity of 1-(piperidin-4-ylcarbonyl)-4-aryl/alkyl-thiosemicarbazides (3–8), there are some SAR trends that were evident. With respect to the substitution on the phenyl ring of lead (3), addition of electron withdrawing groups 4-iodo (5) and 4-bromo (6), and an electron donating group 4-methoxy (6) was well tolerated; all three compounds (4–6) were found to be more active in relation to lead (3). This observation seems to indicate that para-substitution is well tolerated in this chemical series. This hypothesis is further supported by the cytotoxic results for benzyl (7) and ethyl (8) derivatives; these alkyl derivatives were at least 1.25-fold less potent than 3.
Table 3. SAR parameters for compounds 1–8.
In order to clarify the effect of steric and electronic properties of substituents on cytotoxic potency of tested 4-arylthiosemicarbazides (4–6), some parameters such as the bulk parameter based on molar refraction MR and the positionally weighted electronic parameters F and R were taken from the literatureCitation22 and correlated with biological activity. Experiments have shownCitation23,Citation24 that a parameter representing the volume of the substituents on each compound, relative to other members of the same series, may often be linearly correlated with the biological response. As seen in , the most potent aryl analog (5) presents the highest value for MR. Moreover, the compound presents the highest value for the distributive parameter π, which is considered as the parameter of choice for correlating both binding to biological macromolecules and transport through a biological systemCitation25–28. The cytotoxic potency of 5 is probably affected by these descriptors, which can be important parameters for the interaction of 5 with the active sites of topoisomerase(s). Another important results taken from our SAR considerations is that within series of piperidine-thiosemicarbazides (3–8), the most potent 4-iodo analog (5) presents the highest lipophilic character, the highest values for such descriptors as Rf and α, and the lowest value for LUMO while slightly active ethyl derivative (8) presents the lowest values for log P, Rf, and α and the highest value for LUMO suggesting the importance of these molecular parameters on cytotoxic response of studied piperidine-thiosemicarbazides.
Conclusions
As a part of our research for novel antitumor agents, we designed a series of two 4-benzoylthiosemicarbazides (1 and 2) and six piperidine-thiosemicarbazides (3–8). A number of novel thiosemicarbazide derivatives were examined for cytotoxicity in breast cancer cell cultures and for inhibition of topoisomerases I and II. Among the derivatives, 4-benzoylthiosemicarbazides (1 and 2) in both MDA-MB-231 and MCF-7 proved to be only slightly less potent than chlorambucil. In contrast, 4-aryl/alkyl-1-(piperidin-4-yl)-carbonylthiosemicarbazides (3–8) are clearly much less active. Mechanistic studies revealed that 4-benzoylthiosemicarbazides (1 and 2) act as topoisomerase I or topoisomerase II inhibitors in plasmid relaxation assays. The degree to which 4-benzoylthiosemicarbazides (1 and 2)-inhibited cell growth in breast cancer cells was correlated to topoisomerase II-inhibiting activity. The initial results strongly implicate that 4-benzoylthiosemicarbazide scaffold can be developed into efficient chemotherapeutics with dual anticancer and topoisomerase II inhibition activity. For the clear SAR paradigm, however, more compounds should be tested. These studies are underway in our laboratories.
Declaration of interest
This research was supported by Medical University of Bialystok (113-17624F). The authors declare that there are no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Related Research Data
References
- Zhang H, Qian Y, Zhu D, et al. Synthesis, molecular modeling and biological evaluation of chalcone thiosemicarbazide derivatives as novel anticancer agents. Eur J Med Chem 2011;46:4702–8
- Plech T, Wujec M, Siwek A, et al. Synthesis and antimicrobial activity of thiosemicarbazides, s-triazoles and their Mannich bases bearing 3-chlorophenyl moiety. Eur J Med Chem 2011;46:241–8
- Pommier Y, Leo E, Zhang H, Marchand Ch. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol 2010;17:421–33
- Sordet O, Khan QA, Kohn KW, Pommier Y. Apoptosis induced by topoisomerase inhibitors. Curr Med Chem Anti-Cancer Agents 2003;3:271–90
- Larsen AK, Escargueil AE, Skladanowski A. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol Therap 2003;99:167–81
- Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 2002;3:430–40
- René B, Fermandjian S, Mauffret O. Does topoisomerase II specifically recognize and cleave hairpins, cruciforms and crossovers of DNA? Biochimie 2007;89:508–15
- Henry LG, Joseph AP, Mivael O, et al. Topoisomerase II-α as a predictive factor of response to therapy with anthracyclines in locally advanced breast cancer. Breast 2011;20:39–45
- Huang H, Chen Q, Ku X, et al. A series of alpha-heterocyclic carboxaldehyde thiosemicarbazones inhibit topoisomerase II alpha catalytic activity. J Med Chem 2010;53:3048–64
- Rao AV, Klein SR, Agama KK, et al. The iron chelator Dp44mT causes DNA damage and selective inhibition of topoisomerase II alpha in breast cancer cells. Cancer Res 2009;69:948–57
- Moorthy NSHN, Cerqueira, NMFS, Ramos, MJ, Fernandes, PA. Aryl- and heteroaryl-thiosemicarbazones derivatives and its metal complexes: a pharmacological template. Recent Patents on Anticancer Drug Discov 2012;6: [Epub ahead of print]
- Siwek A, Stefańska J. Antimicrobial activity and SAR study of some novel thiosemicarbazide derivatives bearing piperidine moiety. Med Chem 2011;7:690–6
- Siwek A, Staczek P, Stefańska J. Synthesis and structure–activity relationship studies of 4-arylthiosemicarbazides as topoisomerase IV inhibitors with gram-positive antibacterial activity. Search for molecular basis of antibacterial activity of thiosemicarbazides. Eur J Med Chem 2011;46:5717–26
- Carmichael J, Degraff W, Gazdar A, et al. Evaluation of a tetrazolinium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res 1987;47:936–42
- HyperChem 8.0.3, HyperCube Inc., Gainsville, FL, USA, 2007
- Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 1996;118:11225–36
- Jorgensen WL, McDonald NA. Development of an all-atom force field for heterocycles. Properties of liquid pyridine and diazenes. J Mol Struct (THEOCHEM) 1998;424:145–55
- Rocha GB, Freire RO, Simas AM, Stewart JJP. Rm1: a reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I. J Comput Chem 2006;27:1101–11
- Tse WC, Boger DL. Sequence-selective DNA recognition: natural products and nature’s lessons. Chem Biol 2004;11:1607–17
- Neidle S. DNA minor-groove recognition by small molecules. Nat Prod Rep 2001 18:291–309
- Søe K, Rockstroh A, Schache P, Grosse F. The human topoisomerase I damage response plays a role in apoptosis. DNA Repair 2004;3:387–93
- Norrington FE, Hyde RM, Williams SG, Wootton R. Physiochemical-activity relations in practice. 1. A rational and self-consistent data bank. J Med Chem 1975;18:604–7
- Cammarata A. Quantitated structure-activity relationships. Annu Rep Med Chem 1971;6:245–53
- Kutter E, Hansch C. Steric parameters in drug design. Monoamine oxidase inhibitors and antihistamines. J Med Chem 1969;12:647–52
- Fujita T, Iwasa J, Hansch C. A new substituent constant, π, derived from partition coefficients. J Am. Chem Soc 1964;86:5175–80
- Hansch C, Dunn WJ. Linear relationship between lipophilic character and biological activity of drugs. J Pharm Sci 1972;61:1–19
- Hansch C, Clayton JM. Lipophilic character and biological activity of drugs II: the parabolic case. J Pharm Sci 1073;62:1–21
- Bird AE, Marshall AC. Correlation of serum binding of penicillins with partition coefficients. Biochem Pharmacol 1967;16:2275–90