
Abstract
Previously, 2-alkylchromans have been introduced as non-azole inhibitors of 14α-demethylase. Accordingly, we incorporated imidazole ring on the 3-position of 2-alkylchromanones to design new inhibitors of 14α-demethylase and potential antifungal agents. Thus, a series of 2-alkyl-3-imidazolylchromanones were synthesized starting from 2-hydroxyphenacyl bromide. The trans-configuration of compounds was confirmed by NMR-spectroscopy. The antifungal activity of title compounds were evaluated against different fungi in comparison with fluconazole and miconazole. trans-2-(1-Pentyl)-3-imidazolylchroman-4-one (4d) showed the most potent activity against yeasts comparable to fluconazole. The experimental data based on 1H NMR spectroscopy revealed that 2-alkyl side chain and 3-imidazolyl moiety in compound 4d exist predominantly in the di-equatorial conformation. While docking study with 14α-demethylase demonstrated that the di-axial form of compound 4d can be considered as active conformation.
Introduction
In the recent decades, the risk of fungal infections such as candidiasis and aspergillosis has increased greatly because of the rising number of patients who are severely immunocompromised. Thus, invasive fungal infections have become a significant cause of morbidity and mortality in immunocompromised patients suffering from tuberculosis, diabetes, cancer, AIDS and patients who have undergone organ or bone marrow transplantCitation1,Citation2. Moreover, the development of severe resistance is becoming a major problem in the field of antifungal chemotherapyCitation3. On the other hand, there are only a few chemotherapeutic agents that can be used for life-threatening fungal infections. Natural products such as polyenes and echinocandins, and synthetic chemicals including flucytosine and azoles are currently available classes of antifungal agentsCitation4. Among the different classes of antifungal agents that have been discovered, azoles are first-line drugs for the treatment of invasive fungal infections, because of their high therapeutic index. The azoles are chemically either an imidazole or a triazole ring attached to a functionalized phenethyl moiety as their pharmacophore (, structure A). Several new azole antifungal agents, such as luliconazole, voriconazole, pramiconazole, posaconazole, albaconazole and isavuconazole are marketed or currently in the late stages of clinical trialsCitation4,Citation5. However, the extensive use of azoles has led to the development of fungal resistance which significantly reduced their efficacy. Therefore, there is a need to an ongoing search for novel antifungal azoles.
Figure 1. The common structure of azole antifungals (structure A), previously described 2-alkylchromans as non-azole inhibitors for 14α-demethylase of fungi (structure B)Citation9, and 2-alkyl-3-imidazolylchromanones designed as new inhibitors of 14α-demethylase and potential antifungal agents (structure C).
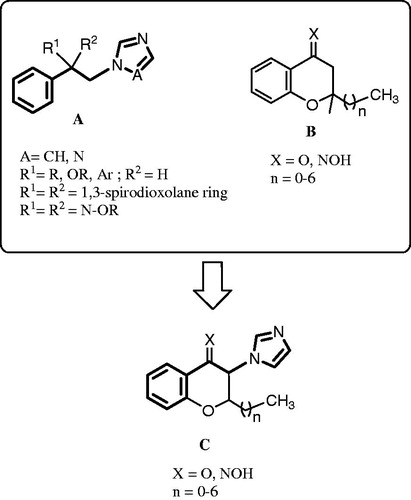
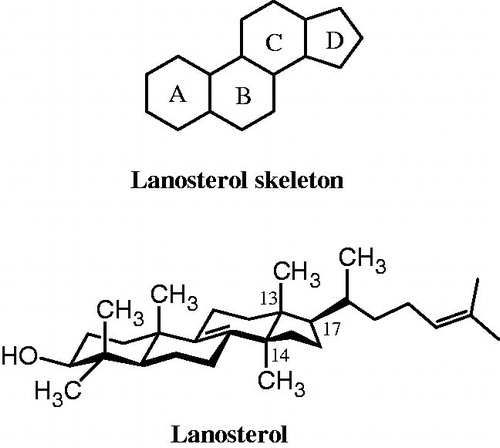
Azole antifungals act by blocking the active site of lanosterol 14α-demethylase (CYP51), a key enzyme in the fungal ergosterol biosynthesisCitation6. The affinity of azole antifungals to the lanosterol 14α-demethylase is not only determined by the coordination binding of the nitrogen of azole ring to the heme iron, but also by the affinity of N-l substituent for the apoprotein part of the enzymeCitation7. The remaining part of the azole antifungal fits in the similar way like lanosterol in the hydrophobic groove of lanosterol 14α-demethylaseCitation8. Previously, Ji et al. designed 2-alkylchromanone and their oximes (, structure B) as non-azole inhibitors of 14α-demethylaseCitation9. Moreover, we have reported 3-imidazolyl chromanone oxime ethers as antifungal agentsCitation10. In this study, we incorporated imidazole ring on the 3-position of 2-alkylchromanones to design new inhibitors of 14α-demethylase and potential antifungal agents (, structure C). It is speculated that the chroman ring having the 2-alkyl side chain in compounds C might be a mimetic of B and C rings or D ring and 17-alkyl side chain of the lanosterol (), and that the 3-imidazolyl residue of C fills the position of the 14-methyl group of lanosterol for coordination binding to the heme of lanosterol 14α-demethylase. Thus, we describe here, synthesis, antifungal activity and docking studies of 2-alkyl-3-imidazolylchromanone derivatives.
Figure 2. The structure of lanosterol; natural substrate of lanosterol 14α-demethylase.
Experimental
Chemistry
The 1-(2-hydroxyphenyl)-2-(1H-imidazol-1-yl)ethanone (2) and trans-2,3-dihydro-3-(1H-imidazol-1-yl)-2-methyl-4H-1-benzopyran-4-one (4a) were prepared according to the literature methodsCitation11,Citation12. The progress of reactions and the purity of compounds were checked by a thin-layer chromatography using Merck silica gel 60 F254 plates (Darmstadt, Germany), and visualization was achieved with UV light (254 nm). Yields are based on purified material and were not optimized. Melting points were determined in open glass capillaries using the Bibby Stuart Scientific SMP3 apparatus (Stuart Scientific, Stone, UK) and are uncorrected. The 1H NMR spectra were recorded using a Bruker 400 or 500 spectrometers (Rheinstatten, Germany), and chemical shifts are expressed as δ (ppm) with tetramethylsilane as internal standard. The spin multiplicities are reported as s (singlet), br s (broad singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). Coupling constants (J) are reported in Hertz (Hz). The IR spectra were obtained on a PerkinElmer (Norwalk, CT) FT-IR spectrophotometer (potassium bromide disks). Elemental analyses were carried out on a CHN-O rapid elemental analyzer (Heraeus GmbH, Hanau, Germany) for C, H and N, and the results are within ±0.4% of the theoretical values.
General procedure for the synthesis of trans-2,3-dihydro-3-(1H-imidazol-1-yl)-2-alkyl-4H-1-benzopyran-4-one nitrate (4b–g)
A solution of compound 2 (404 mg, 2.0 mmol) and appropriate aliphatic aldehyde (2.4 mmol) in 2-propanol (4 mL) containing piperidine (0.67 mmol) was heated under reflux for 2–4 h. The reaction was concentrated under reduced pressure and mixed with water (20 mL). The mixture was left in the fridge overnight until viscose oil was separated. The aqueous phase was decanted and the oily residue was dissolved in diethyl ether, and treated with 65% HNO3 (0.2 mL). The mixture was cooled at a freezer to complete the precipitation of the nitrate salts 4b–g, which were collected by filtration and washed with diethyl ether.
trans-2,3-Dihydro-3-(1H-imidazol-1-yl)-2-(1-propyl)-4H-1-benzopyran-4-one nitrate (4b)
Yield 25%; m.p. 111 °C–112 °C; IR (KBr, cm−1) νmax: 1715 (C=O). 1H NMR (500 MHz, DMSO-d6) δ: 0.86 (t, J = 7.25 Hz, 3H, CH3), 1.22–1.46 (m, 2H, CH2), 1.51–1.77 (m, 2H, CH2), 5.13 (m, 1H, H-2), 6.00 (d, J = 12.35 Hz, 1H, H-3), 7.19 (d, J = 8.1 Hz, 1H, H-8), 7.20 (t, J = 7.8 Hz, 1H, H-6), 7.71 (t, J = 7.3 Hz, 1H, H-7), 7.78–7.91 (m, 2H, imidazole), 7.84 (d, J = 6.75 Hz, 1H, H-5), 9.22 (br s, 1H, imidazole). MS (m/z, %): 256 (M+, 60), 255 (77), 213 (34), 188 (35), 173 (20), 150 (32), 136 (47), 121 (85), 120 (100), 109 (68), 107 (53), 92 (68), 80 (26), 71 (47), 69 (96), 53 (39). Anal. Calcd for C15H17N3O5 (%): C, 56.42; H, 5.37; N, 13.16. Found (%): C, 56.70; H, 5.59; N, 13.12.
trans-2,3-Dihydro-3-(1H-imidazol-1-yl)-2-(1-butyl)-4H-1-benzopyran-4-one nitrate (4c)
Yield 52%; m.p. 108 °C–109 °C; IR (KBr, cm−1) νmax: 1714 (C=O). 1H NMR (500 MHz, DMSO-d6) δ: 0.84 (t, J = 7.2 Hz, 3H, CH3), 1.19–1.42 (m, 4H, CH2CH2), 1.48–1.61 (m, 1H, HCHa), 1.62–1.74 (m, 1H, HCHb), 5.13 (m, 1H, H-2), 6.00 (d, J = 12.35 Hz, 1H, H-3), 7.19 (d, J = 8.0 Hz, 1H, H-8), 7.20 (t, J = 6.7 Hz, 1H, H-6), 7.71 (dt, J = 7.78 and 1.55 Hz, 1H, H-7), 7.84 (m, 3H, two imidazole-H and H-5), 9.22 (s, 1H, imidazole). MS (m/z, %): 270 (M+, 15), 269 (55), 213 (23), 189 (35), 176 (55), 150 (100), 121 (96), 107 (19), 92 (57), 85 (45), 68 (45), 57 (51). Anal. Calcd for C16H19N3O5 (%): C, 57.65; H, 5.75; N, 12.61. Found (%): C, 57.58; H, 5.87; N, 12.33.
trans-2,3-Dihydro-3-(1H-imidazol-1-yl)-2-(1-pentyl)-4H-1-benzopyran-4-one nitrate (4d)
Yield 35%; m.p. 124 °C–125 °C; IR (KBr, cm−1) νmax: 1715 (C=O). 1H NMR (500 MHz, DMSO-d6) δ: 0.85 (t, J = 6.2 Hz, 3H, CH3), 1.14–1.42 (m, 6H, CH2CH2CH2), 1.50–1.62 (m, 1H, HCHa), 1.64–1.75 (m, 1H, HCHb), 5.13 (m, 1H, H-2), 6.00 (d, J = 12.3 Hz, 1H, H-3), 7.19 (d, J = 8.35 Hz, 1H, H-8), 7.21 (t, J = 8.0 Hz, 1H, H-6), 7.71 (t, J = 7.5 Hz, 1H, H-7), 7.78–7.91 (m, 2H, imidazole), 7.84 (d, J = 8.15 Hz, 1H, H-5), 9.22 (s, 1H, imidazole). MS (m/z, %): 284 (M+, 18), 283 (45), 232 (15), 213 (23), 203 (22), 189 (29), 176 (77), 150 (90), 121 (100), 105 (26), 99 (54), 92 (27), 69 (61), 55 (28). Anal. Calcd for C17H21N3O5 (%): C, 58.78; H, 6.09; N, 12.10. Found (%): C, 59.01; H, 5.88; N, 11.97.
trans-2,3-Dihydro-3-(1H-imidazol-1-yl)-2-(1-hexyl)-4H-1-benzopyran-4-one nitrate (4e)
Yield 44%; m.p. 112 °C–113 °C; IR (KBr, cm−1) νmax: 1708 (C=O). 1H NMR (400 MHz, DMSO-d6) δ: 0.86 (t, J = 6.8 Hz, 3H, CH3), 1.10–1.78 (m, 10H, CH2CH2CH2CH2CH2), 5.12 (m, 1H, H-2), 5.98 (d, J = 12 Hz, 1H, H-3), 6.82 (br s, 1H, imidazole), 7.17 (d, J = 8.0 Hz, 1H, H-8), 7.20 (dt, J = 7.60 and 0.8 Hz, 1H, H-6), 7.35 (br s, 1H, imidazole), 7.71 (dt, J = 8.6 and 1.6 Hz, 1H, H-7), 7.84 (dd, J = 7.8 and 1.6 Hz, 1H, H-5), 9.23 (s, 1H, imidazole). MS (m/z, %): 298 (M+, 25), 297 (70), 213 (42), 189 (18), 176 (37), 150 (76), 121 (100), 113 (57), 107 (25), 92 (31), 85 (26), 68 (55), 55 (25). Anal. Calcd for C18H23N3O5 (%): C, 59.82; H, 6.41; N, 11.63. Found (%): C, 59.77; H, 6.48; N, 11.84.
trans-2,3-Dihydro-3-(1H-imidazol-1-yl)-2-(1-heptyl)-4H-1-benzopyran-4-one nitrate (4f)
Yield 28%; m.p. 95 °C–96 °C; IR (KBr, cm−1) νmax: 1715 (C=O). 1H NMR (500 MHz, DMSO-d6) δ: 0.85 (t, J = 6.88 Hz, 3H, CH3), 1.15–1.40 (m, 10H, CH2CH2CH2CH2CH2), 1.48–1.62 (m, 1H, HCHa), 1.63–1.78 (m, 1H, HCHb), 5.13 (m, 1H, H-2), 5.99 (d, J = 12.35 Hz, 1H, H-3), 7.19 (d, J = 8.4 Hz, 1H, H-8), 7.20 (t, J = 7.53 Hz, 1H, H-6), 7.71 (dt, J = 7.8 and 1.60 Hz, 1H, H-7), 7.77–7.85 (m, 1H, H-5 and 2H, imidazole), 9.20 (s, 1H, imidazole). MS (m/z, %): 312 (M+, 25), 311 (64), 297 (27), 213 (42), 189 (15), 176 (29), 150 (30), 127 (53), 121 (63), 107 (18), 92 (25), 69 (96), 57 (100). Anal. Calcd for C19H25N3O5 (%): C, 60.79; H, 6.71; N, 11.19. Found (%): C, 60.88; H, 6.49; N, 10.89.
trans-2,3-Dihydro-3-(1H-imidazol-1-yl)-2-(1-isobutyl)-4H-1-benzopyran-4-one nitrate (4g)
Yield 40%; m.p. 100 °C–101 °C; IR (KBr, cm−1) νmax: 1705 (C=O). 1H NMR (500 MHz, DMSO-d6) δ: 0.83 (d, J = 6.5 Hz, 3H, CH3), 0.85–0.95 (m, 1H, CH(Me)2), 0.89 (d, J = 6.6 Hz, 3H, CH3), 1.78–1.96 (m, 2H, CH2), 5.21 (m, 1H, H-2), 5.97 (d, J = 12.2 Hz, 1H, H-3), 7.19 (d, J = 8.3 Hz, 1H, H-8), 7.20 (t, J = 7.4 and 1.0 Hz, 1H, H-6), 7.71 (dt, J = 7.78 and 1.7 Hz, 1H, H-7), 7.76–7.88 (m, 2H, imidazole), 7.84 (dd, J = 7.8 and 1.5 Hz, 1H, H-5), 9.17 (br s, 1H, imidazole). MS (m/z, %): 270 (M+, 35), 269 (100), 213 (67), 202 (27), 187 (32), 150 (35), 121 (62), 107 (53), 92 (36), 69 (46), 57 (50). Anal. Calcd for C16H19N3O5 (%): C, 57.65; H, 5.75; N, 12.61. Found (%): C, 57.37; H, 5.81; N, 12.73.
General procedure for the synthesis of (Z)-trans-2,3-dihydro-3-(1H-imidazol-1-yl)-2-alkyl-4H-1-benzopyran-4-one oximes (5a–g)
A solution of compound 2 (404 mg, 2.0 mmol) and appropriate aliphatic aldehyde (2.4 mmol) in 2-propanol (4 mL) containing piperidine (0.67 mmol) was heated under reflux for 2–4 h. The reaction was concentrated under reduced pressure and mixed with water (20 mL). The mixture was left in the fridge overnight until viscose oil was separated. The aqueous phase was decanted and the oily residue was dissolved in methanol (3 mL). After an addition of hydroxylamine hydrochloride (417 mg, 6 mmol) and K2CO3 (276 mg, 2.0 mmol), the mixture was stirred at room temperature for 2–6 d. Then, the reaction mixture was poured into water, and the precipitate was filtered, washed with water to give oxime derivative 5.
(Z)-trans-2,3-Dihydro-3-(1H-imidazol-1-yl)-2-ethyl-4H-1-benzopyran-4-one oxime (5a)
Yield 58%; m.p. 160–162 °C; IR (KBr, cm−1) νmax: 3450 (O–H), 1605 (C=NOH). 1H NMR (500 MHz, DMSO-d6) δ: 0.98 (t, J = 7.4 Hz, 3H, CH3), 1.17–1.29 (m, 1H, HCHa), 1.41–1.52 (m, 1H, HCHb), 4.19 (m, 1H, H-2), 5.83 (d, J = 1.1 Hz, H-3), 6.85 (br s, 2H, H-4 and H-5 imidazole), 7.04 (d, J = 8.35 Hz, 1H, H-8), 7.06 (t, J = 7.85 Hz, 1H, H-6), 7.37 (dt, J = 7.75 and 1.5 Hz, 1H, H-7), 7.61 (br s, 1H, imidazole H), 7.84 (dd, J = 7.85 and 1.5 Hz, 1H, H-5), 11.82 (s, 1H, OH). MS (m/z, %): 257 (M+, 100), 240 (18), 213 (18), 189 (48), 173 (32), 146 (30), 132 (25), 121 (53), 102 (19), 91 (26), 69 (34), 53 (26). Anal. Calcd for C14H15N3O2 (%): C, 65.35; H, 5.88; N, 16.33. Found (%): C, 65.33; H, 6.02; N, 16.40.
(Z)-trans-2,3-Dihydro-3-(1H-imidazol-1-yl)-2-(1-propyl)-4H-1-benzopyran-4-one oxime (5b)
Yield 40%; m.p. 184 °C–185 °C; IR (KBr, cm−1) νmax: 3450 (O–H), 1635 (C=NOH). 1H NMR (500 MHz, DMSO-d6) δ: 0.87 (t, J = 6.8 Hz, 3H, CH3), 1.10–1.65 (m, 4H, CH2CH2), 4.23–4.30 (m, 1H, H-2), 5.80 (d, J = 1.2 Hz, 1H, H-3), 6.81 (br s, 1H, imidazole), 6.83 (br s, 1H, imidazole), 7.03 (d, J = 8.1 Hz, 1H, H-8), 7.06 (t, J = 7.6 Hz, 1H, H-6), 7.37 (t, J = 7.7 Hz, 1H, H-7), 7.56 (br s, 1H, imidazole), 7.84 (d, J = 7.75 Hz, 1H, H-5), 11.80 (s, 1H, OH). MS (m/z, %): 271 (M+, 100), 254 (76), 227 (20), 203 (38), 186 (21), 174 (19), 159 (19), 146 (35), 131 (20), 120 (28), 91 (18), 69 (28), 53 (20). Anal. Calcd for C15H17N3O2 (%): C, 66.40; H, 6.32; N, 15.49. Found (%): C, 66.34; H, 6.39; N, 15.80.
(Z)-2,3-Dihydro-3-(1H-imidazol-1-yl)-2-(1-butyl)-4H-1-benzopyran-4-one oxime (5c)
Yield 44 %; m.p. 142 °C–144 °C; IR (KBr, cm−1) νmax: 3450 (O–H), 1606 (C=NOH). 1H NMR (500 MHz, DMSO-d6) δ: 0.85 (t, J = 7.5 Hz, 3H, CH3), 1.12–1.60 (m, 6H, CH2CH2CH2), 4.24 (m, 1H, H-2), 5.81 (d, J = 1.2 Hz, H-3), 6.83 (br s, 1H, imidazole), 6.85 (br s, 1H, imidazole), 7.03 (d, J = 7.5 Hz, H-8), 7.05 (dt, J = 7.5 and 1.0 Hz, H-6), 7.62 (br s, 1H, imidazole), 7.84 (dd, J = 8.0 and 1.5 Hz, H-5), 11.83 (s, 1H, OH). MS (m/z, %): 285 (M+, 100), 268 (87), 241 (19), 228 (20), 217 (38), 200 (22), 174 (28), 159 (30), 146 (32), 131 (23), 120 (32), 107 (24), 91 (26), 81 (24), 69 (49), 53 (28). Anal. Calcd for C16H19N3O2 (%): C, 67.35; H, 6.71; N, 14.73. Found (%): C, 67.66; H, 6.53; N, 14.64.
(Z)-trans-2,3-Dihydro-3-(1H-imidazol-1-yl)-2-(1-pentyl)-4H-1-benzopyran-4-one oxime (5d)
Yield 86%; m.p. 150 °C–152 °C; IR (KBr, cm−1) νmax: 3450 (O–H), 1605 (C=NOH). 1H NMR (400 MHz, DMSO-d6) δ: 0.85 (t, J = 7.0 Hz, 3H, CH3), 0.98–1.62 (m, 8H, CH2CH2CH2CH2), 4.22 (m, 1H, H-2), 5.81 (m, 1H, H-3), 6.82 (br s, 2H, H-4 and H-5 imidazole), 7.03 (d, J = 8.4 Hz, 1H, H-8), 7.05 (t, J = 7.6 Hz, 1H, H-6), 7.37 (dt, J = 7.2 and 1.2 Hz, 1H, H-7), 7.57 (m, 1H, imidazole), 7.84 (d, J = 8.0 Hz, 1H, H-5), 11.80 (s, 1H, OH). MS (m/z, %): 299 (M+, 89), 282 (100), 255 (24), 231 (29), 212 (24), 188 (20), 174 (26), 159 (29), 146 (30), 131 (23), 120 (35), 107 (24), 91 (18), 81 (23), 69 (45), 55 (27). Anal. Calcd for C17H21N3O2 (%): C, 68.20; H, 7.07; N, 14.04. Found (%): C, 68.11; H, 7.20; N, 14.02.
(Z)-trans-2,3-Dihydro-3-(1H-imidazol-1-yl)-2-(1-hexyl)-4H-1-benzopyran-4-one oxime (5e)
Yield 87%; m.p. 158 °C–159 °C; IR (KBr, cm−1) νmax: 3450 (O–H), 1622 (C=NOH). 1H NMR (500 MHz, DMSO-d6) δ: 0.86 (t, J = 6.8 Hz, 3H, CH3), 1.17–1.57 (m, 10H, CH2CH2CH2CH2CH2), 4.25 (m, 1H, H-2), 5.81 (d, J = 2.15 Hz, 1H, H-3), 6.82 (br s, 1H, imidazole), 6.84 (br s, 1H, imidazole), 7.04 (d, J = 8.15 Hz, 1H, H-8), 7.07 (t, J = 7.2 Hz, 1H, H-6), 7.38 (dt, J = 7.75 and 1.60 Hz, 1H, H-7), 7.57 (br s, 1H, imidazole), 7.85 (dd, J = 7.9 and 1.45 Hz, 1H, H-5), 11.82 (s, 1H, OH). MS (m/z, %): 313 (M+, 64), 296 (100), 269 (21), 245 (22), 228 (32), 212 (30), 188 (19), 174 (21), 159 (31), 146 (32), 131 (32), 121 (41), 107 (31), 91 (30), 81 (26), 69 (58), 55 (42). Anal. Calcd for C18H23N3O2 (%): C, 68.98; H, 7.40; N, 13.41. Found (%): C, 69.07; H, 7.22; N, 13.47.
(Z)-trans-2,3-Dihydro-3-(1H-imidazol-1-yl)-2-(1-heptyl)-4H-1-benzopyran-4-one oxime (5f)
Yield 63%; m.p. 147 °C–148 °C; IR (KBr, cm−1) νmax: 3460 (O–H), 1604 (C=NOH). 1H NMR (400 MHz, DMSO-d6) δ: 0.62–1.40 (m, 14H, HCHaCH2CH2CH2CH2CH2CH3), 1.80–1.97 (m, 1H, HCHb), 4.31–4.47 (m, 1H, H-2), 5.75–5.94 (m, 1H, H-3), 6.85 (br s, 2H, H-4 and H-5 imidazole), 7.03 (d, J = 8.8 Hz, 1H, H-8), 7.06 (t, J = 7.6 Hz, 1H, H-6), 7.37 (t, J = 7.6 Hz, 1H, H-7), 7.61 (br s, 1H, imidazole), 7.85 (d, J = 8.0 Hz, 1H, H-5), 11.82 (s, 1H, OH). MS (m/z, %): 327 (M+, 44), 310 (61), 283 (15), 259 (34), 228 (19), 212 (61), 188 (20), 173 (24), 160 (22), 145 (36), 127 (43), 121 (54), 107 (29), 92 (27), 82 (36), 69 (100), 55 (46). Anal. Calcd for C19H25N3O2 (%): C, 69.70; H, 7.70; N, 12.83. Found (%): C, 69.72; H, 7.59; N, 12.97.
(Z)-trans-2,3-Dihydro-3-(1H-imidazol-1-yl)-2-(1-isobutyl)-4H-1-benzopyran-4-one oxime (5g)
Yield 50%; m.p. 169 °C–171 °C; IR (KBr, cm−1) νmax: 3460 (O–H), 1606 (C=NOH). 1H NMR (400 MHz, DMSO-d6) δ: 0.62–1.80 (m, 9H, CH2CH(CH3)2), 4.18–4.28 (m, 1H, H-2), 5.76–5.84 (m, 1H, H-3), 6.80 (br s, 1H, imidazole), 6.82 (br s, 1H, imidazole), 7.02 (d, J = 8.0 Hz, 1H, H-8), 7.05 (t, J = 7.6 Hz, 1H, H-6), 7.37 (t, J = 7.4 Hz, 1H, H-7), 7.56 (br s, 1H, imidazole), 7.84 (d, J = 7.6 Hz, 1H, H-5), 11.82 (s, 1H, OH). MS (m/z, %): 285 (M+, 62), 268 (100), 228 (16), 217 (20), 212 (25), 200 (17), 184 (17), 174 (24), 161 (25), 146 (34), 131 (22), 120 (21), 107 (13), 91 (18), 81 (15), 69 (43), 53 (18). Anal. Calcd for C16H19N3O2 (%): C, 67.35; H, 6.71; N, 14.73. Found (%): C, 67.51; H, 6.70; N, 14.44.
Antifungal activity assay
The minimum inhibitory concentrations (MICs) of compounds against Candida albicans ATCC 10231, Saccharomyces cerevisiae PTCC 5177, Aspergillus niger PTCC 5011 and Microsporum gypseum PTCC 5070 were determined by micro-dilution methodCitation13. A stock solution (128 µg/mL) of compounds was prepared in a Sabouraud dextrose broth (SDB) by using 10% v/v DMSO. The stock solution of compounds was transferred into the plates with 96 U-shaped wells and serially diluted with SDB in subsequent wells. Then, fungal suspension was added to each well to reach the final inoculum size of 0.5 × 103 CFU/mL. The final concentrations of tested compounds in the medium were 64, 32, 16, 8, 4, 2, 1 and 0.5 µg/mL. Fungal plates were made in triplicate and incubated at 35 °C about 24–48 h for C. albicans and S. cerevisiae, about 72 h for A. niger and about 96 h for M. gypseum. The MIC value was defined as lowest concentration of the antifungal agent at which there were no visible growth shown.
Computational method
Crystal structure of cytochrome P450 14α-sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with fluconazole with ID 1EA1 was retrieved from the Brookhaven Protein Data Bank (http://www.rcsb.org). The FlexX program interfaced with LeadIT 2.0.2 (BioSolveIT GmbH, Sankt Augustin, Germany) was used for docking. The binding site was defined to any residues 8 Å distant from the co-crystallized fluconazole in the complex crystal structure of the enzyme. For an evaluation of the ligands affinity towards active site, the HYDE assessment module of LeadIT software was used to report the free energy of binding (ΔG) and ligand efficiency of the best scored poseCitation14,Citation15.
Results and discussion
Chemistry
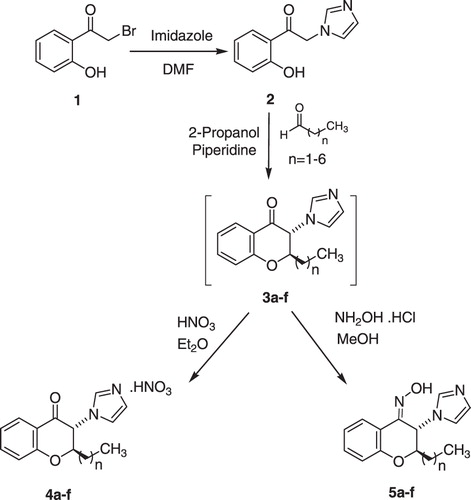
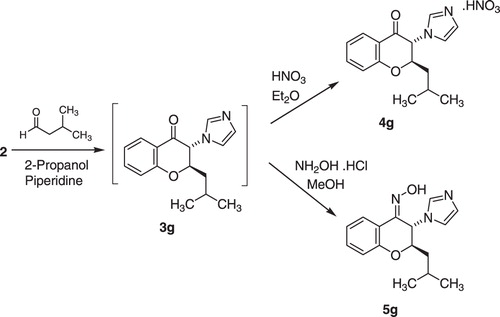
The designed 2-alkyl-3-imidazolylchromanones were synthesized starting from 2-hydroxyphenacyl bromide (1) as outlined in . Compound 1 was reacted with excess imidazole in DMF to give N-(2-hydroxyphenacyl)imidazole derivative 2Citation11. Cyclization of compound 2 with linear aliphatic aldehydes occurred in refluxing 2-propanol, in the presence of piperidine as a catalyst. The oily crude product (3) was trans-stereoisomer, which underwent nitrate salt formation by using nitric acid in diethyl ether. The 2-alkyl-3-imidazolylchromanone nitrate (4) was obtained as a pure crystalline product. For preparation of oxime derivatives 5, the crude product (3) was treated with excess hydroxylamine hydrochloride in methanol at room temperature. In this reaction, the addition of K2CO3 assisted to the reaction completion. The oxime product (5) was assigned as (Z)-trans-stereoisomer. As shown in , the isobutyl derivative (3g) was prepared by the reaction of intermediate 2 and 3-methylbutanal. Further reaction steps were the same as described for compounds 4a–f and 5a–f.
Scheme 1. Synthesis of chromanone derivatives (4a–f) and chromanone (Z)-oxime (5a–f).
Scheme 2. Synthesis of compounds 4g and 5g.
General and specific aspects of compounds' structures were elucidated by 1H NMR spectroscopy. represents the chemical shifts of the core structure based on the protons of chroman and imidazole rings. Since the N-3 atom of imidazole ring is protonated in the nitrate salts of compounds 4, the chemical shift of proton at the C-2 position of imidazole ring is deshielded. The large vicinal coupling constant (J2,3 = 12.0–12.35 Hz) between H-2 and H-3 of chroman ring indicates that the orientation of the 2-alkyl group and the 3-imidazole ring is trans. In the 1H NMR spectra of oxime derivatives 5, only a set of signals were appeared which confirmed the exclusive formation of one stereoisomer during the oximation reaction. The (E)- or (Z)-geometry was readily identified by 1H NMR, based on the deshielding effect of the oxime oxygen. According to the previous studies, when the (E)-isomer is produced in the oximation reaction, the H-5 proton of chroman ring is significantly deshielded (δ > 8.5 ppm) with respect to the parent ketone (δ < 8 ppm)Citation10,Citation16. As seen in , the chemical shifts of oxime derivatives (5) are virtually the same as those of ketones (4) and no signal was detected around 8.5 ppm. Thus, the (E)-configuration was consequently ruled out and the orientation of the oxime moiety of compound 5 was assigned to be of (Z)-geometry.
Table 1. The chemical shifts of compounds 4 and 5.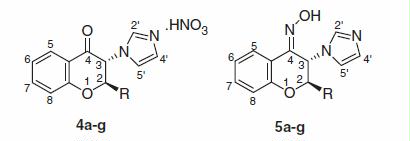
It should be noted that all designed compounds 4 and 5 possess two chiral centers in their chroman core structure at the C-2 and C-3 positions. These compounds are actually as racemates but for more clarity, only one enantiomer was presented. Similarly, many of marketed azole antifungals such as miconazole, bifonazole, sertaconazole, ketoconazole, itraconazole and terconazole are racemic mixtures. In the 1H NMR spectra of compounds 4 and 5, the signals of methylene group which connected to the chroman ring indicated the diastereotopic nature of the methylene group protons because of the presence of C-2 stereocenter in chroman ring.
Antifungal activity
The antifungal activity of designed 2-alkyl-3-imidazolylchroman derivatives 4a–g and 5a–g were evaluated by determining their MICs against C. albicans (yeast), S. cerevisiae (yeast), M. gypseum (dermatophyte) and A. niger (mould). The MICs of compounds were determined by the micro-dilution method in comparison with fluconazole and miconazole as standard azole antifungal agents. The results were summarized in .
Table 2. Antifungal activity (MICs, µg/mL) of compounds 4a–g and 5a–g in comparison with standard antifungal drugs.
The MIC values of title compounds in comparison with those of fluconazole and miconazole were presented in . In the case of C. albicans and S. cerevisiae, compound 4d was the most potent compound with MIC values of 8 µg/mL. Its growth inhibitory activity against yeasts was comparable to fluconazole. Also, compound 4f showed good inhibitory activity (MICs = 8–16 µg/mL) against yeasts comparable to fluconazole. Furthermore, compounds 4c, 4e, 5c and 5d were active against yeasts with MIC values of 16–64 µg/mL. Although, fluconazole was inactive against M. gypseum and A. niger (MICs > 64 µg/mL), but compounds 4d–f, 5a, 5c–e and 5g exhibited mild activity against these fungi. A survey of MIC values in revealed that the imidazolylchromanones (4a–c and 4g) containing small side chain (1C–4C) were inactive. While chromanones bearing longer 2-alkyl group (>4C) showed better antifungal activity against all fungi strains tested. The comparison of ketone compounds (4) with related oxime derivatives (5) demonstrated that oximation had negative effect and diminished the antifungal activity. Also, compounds 4g and 5g with a branched alkyl group (isobutyl) showed no activity against C. albicans, S. cerevisiae and A. niger.
Docking study
With the aim of rationalizing the antifungal activity data obtained, docking study was performed for the selected derivatives 4d (keto-compound) and its oxime analog 5d, in order to investigate the possible interactions with Cytochrome P450 14α-sterol demethylase. Since no experimental data were available for the target enzyme Candida P450 DM in the Protein Data Bank, the crystallographic structure of the complex between Cytochrome P450 14α-sterol demethylase from M. tuberculosis (Mycobacterium P450 DM) and fluconazole with the ID 1EA1 was used for the docking study.
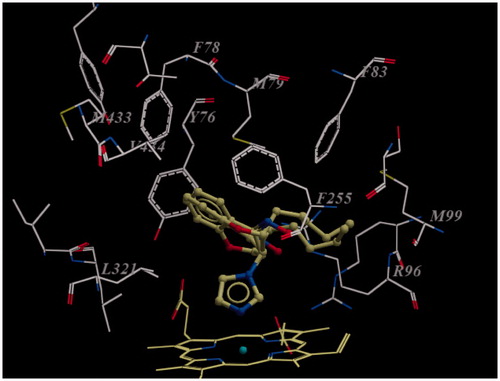
As depicted in , both (SS)- and (RR)-enantiomers of compounds 4d and 5d were included for docking studies. The aromatic part of chroman ring in both compounds 4d and 5d is situated in a lipophilic region and interacts with Phe255, Met79, Tyr76, Val434, Leu321 and Phe78. The pentyl side chain is in close contact with Phe83, Leu100 and Ala256. Furthermore, the imidazole ring of both compounds is positioned above the porphyrine plane, in which the N-3 atom of imidazole ring is coordinated to the heme iron. Distance between the nitrogen atom and heme iron is 2.2 Å in both compounds, which is in good agreement with that found in the crystal structure of Mycobacterium P450 DM complexed with fluconazole (2.3 Å) (). By investigating the binding mode of compound 4d enantiomers (), it is revealed that the orientation of pyran ring is different in the two enantiomers. On the other hand, in both enantiomers the aromatic part of chroman ring, pentyl side chain and imidazole ring occupy the same place in the active site, but carbonyl groups in both the enantiomers of compound 4d are facing in the opposite directions. However, hydrogen bonding is not disturbed by oppositely oriented carbonyl groups, because Arg96 and His259 are in close vicinity to the respective carbonyl groups for hydrogen bonding in both the RR and SS enantiomeric forms (3.1 Å and 4.8 Å, respectively).
Figure 3. Docking pose view (2D map) for the enantiomers of compounds 4d and 5d. Only important amino acids for interactions are shown.
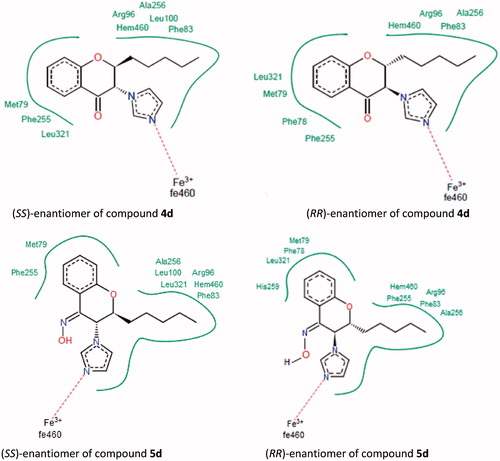
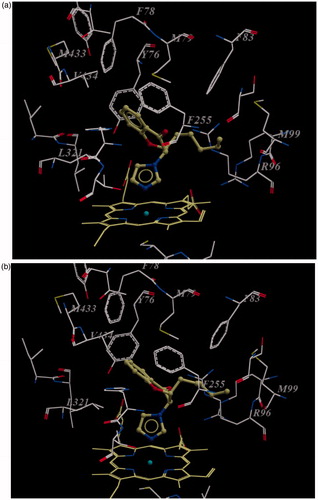
Figure 4. (a) (SS)-enantiomer of compound 4d in the active site of enzyme. Note that carbonyl group of compound 4d orients towards Phe255 carbonyl group; (b) (RR)-enantiomer of compound 4d in the active site of enzyme. Note that carbonyl group of compound 4d orients toward the guanidine group of Arg96. For the sake of clarity only amino acids within 7 Å distant from the docked ligand are shown.
As shown in , the binding mode of the oxime derivative (compound 5d) is highly similar to compound 4d because the same place in the active site has been occupied by the phenyl ring, pentyl side chain and imidazole ring of compound 5d. Moreover, the oxime group has been superimposed on the carbonyl group of the parent compound 4d but hydrogen bonding is different.
Figure 5. (RR)- and (SS)-enantiomers of compound 5d in the active site of enzyme. Note that oxime groups have been oriented in the opposite directions. The oxime group of (RR)-enantiomer orients toward the guanidine group of Arg96, while the oxime group of (SS)-enantiomer orients towards the Phe255 carbonyl group. For both enantiomers, phenyl ring, pentyl side chain and imidazole ring occupy the same place in the active site. For the sake of clarity only amino acids within 7 Å distant from the docked ligand are shown.
For evaluation of the ligands affinity towards active site, the HYDE assessment module of LeadIT software was used to report the free energy of binding (ΔG) and ligand efficiency of the best scored pose (). HYDE is an empirical scoring function, which assesses protein–ligand complex by considering hydrogen bond interactions and also hydrophobic and desolvation effects and provides estimation for the binding affinityCitation14,Citation15. For the (SS)-enantiomeric form of 5d (with ΔGbinding = −42 kJ/mol), hydrogen bonding seems to be more possible between the oxime hydrogen atom and carbonyl oxygen atom of Phe255 (3 Å). In the (RR)-enantiomer (with ΔGbinding = −45 kJ/mol), stronger hydrogen bonding is possible between the oxime group and the guanidine group of Arg96 due to a very close proximity (2 Å) of the two groups (). In both compounds 4d and 5d, (RR)-enantiomers have better interactions in the active site of the enzyme with the lower energy of binding compared to the (SS)-enantiomers. Investigating the binding mode shows that Arg96 plays a key role for hydrogen bonding to the carbonyl or the oxime group of the RR-enantiomer of both compounds 4d and 5d.
Table 3. HYDE scoring reported affinities of ligands toward cytochrome P450 14α-sterol demethylase.
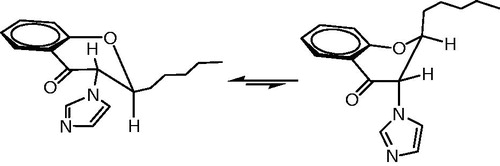
As discussed in the “Chemistry” section, the large vicinal coupling constant between H-2 and H-3 of chroman ring in the compound 4d prototype indicates that the orientation of the 2-alkyl group and the 3-imidazole ring is trans. Theoretically, the fused six-membered hetero-ring of chroman moiety can exist in the form of two energetically different half-chairs in which 2-alkyl side chain and 3-imidazolyl moiety occupy di-axial or di-equatorial positions (). The relatively large J2,3 value in compound 4d prototype confirms the preferred di-axial conformation of H-2 and H-3Citation15. Thus, it is believed that 2-alkyl side chain and 3-imidazolyl moiety in compound 4d exist predominantly in the di-equatorial conformation. FlexX is an automated docking program which considers ligand conformational flexibility by an incremental fragment placing methodCitation17. In our docking study on compound 4d, imidazole ring and pentyl group have adopted a di-axial orientation due to ring flipping after docking calculations. Therefore, the di-axial form which has lower binding energy and better interaction with amino acids in the active site can be considered as active conformation.
Figure 6. Possible conformations for trans-2-(1-pentyl)-3-imidazolylchroman-4-one (4d).
Conclusion
In this study, we incorporated imidazole ring on the 3-position of 2-alkylchromanones to design new inhibitors of 14α-demethylase and potential azole antifungal agents. The designed 2-alkyl-3-imidazolylchromanones were synthesized selectively and assigned as trans-stereoisomer. Furthermore, the (Z)-oxime derivatives of 2-alkyl-3-imidazolylchromanones were prepared exclusively and confirmed their geometries by 1H NMR spectroscopy. The results of antifungal evaluation against different strains of fungi demonstrated that the length of alkyl side chain had a significant impact on antifungal activity. In general, chromanones bearing longer 2-alkyl group (>4C) showed better antifungal activity against all fungi tested. Among the chroman-4-ones, 2-(1-pentyl) derivative (4d) showed the most potent activity against yeasts comparable to fluconazole. Docking study of selected compounds 4d and 5d with cytochrome P450 14α-sterol demethylase revealed that the binding mode of the oxime derivative (compound 5d) is highly similar to parent ketone (compound 4d) because the same place in the active site has been occupied by the phenyl ring, pentyl side chain and imidazole ring of compound 5d. Furthermore, the docking study demonstrated that the di-axial form of target compounds can be considered as active conformation.
Declaration of interest
The authors have declared no conflict of interest.
This work was supported by a grant from the Research Council of Mazandaran University of Medical Sciences, Sari, Iran.
Acknowledgements
A part of this work was related to the Pharm.D thesis of Touba Banipoulad (Faculty of Pharmacy, Mazandaran University of Medical Sciences).
References
- Mishra NN, Prasad T, Sharma N, et al. Pathogenicity and drug resistance in Candida albicans and other yeast species. A review. Acta Microbiol Immunol Hung 2007;54:201–35
- Lai CC, Tan CK, Huang YT, et al. Current challenges in the management of invasive fungal infections. J Infect Chemother 2008;14:77–85
- Okeke IN, Laxminarayan R, Bhutta ZA, et al. Antimicrobial resistance in developing countries. Part I: recent trends and current status. Lancet Infect Dis 2005;5:481–93
- Van Minnebruggen G, François IEJA, Cammue BPA, et al. A general overview on past, present and future antimycotics. Open Mycol J 2010;4:22–32
- Girmenia C. New generation azole antifungals in clinical investigation. Expert Opin Investig Drugs 2009;18:1279–95
- Guardiola HM, Foster L, Mushrush D, Vaz AD. Azole-antifungal binding to a novel cytochrome P450 from Mycobacterium tuberculosis: implications for treatment of tuberculosis. Biochem Pharmacol 2001;61:1463–70
- Yoshida Y, Aoyama Y. Interaction of azole fungicides with yeast cytochrome P-450 which catalyzes lanosterol 14α-demethylation. In: Iwata K, Vanden Bossche H, eds. In vitro and in vivo evaluation of antifungal agents. Amsterdam: Elsevier Science; 1986:123–34
- Aoyama Y, Yoshida Y. The 3-hydroxy group of lanosterol is essential for orienting the substrate site of cytochrome P-450(14DM) (lanosterol 14 alpha-demethylase). Biochim Biophys Acta 1989;1006:209–13
- Ji H, Zhang W, Zhang M, et al. Structure-based de novo design, synthesis, and biological evaluation of non-azole inhibitors specific for lanosterol 14alpha-demethylase of fungi. J Med Chem 2003;46:474–85
- Emami S, Falahati M, Banifatemi A, et al. Stereoselective synthesis and in vitro antifungal evaluation of (E)- and (Z)-imidazolylchromanone oxime ethers. Arch Pharm (Weinheim) 2002;335:318–24
- Emami S, Foroumadi A, Falahati M, et al. 2-Hydroxyphenacyl azoles and related azolium derivatives as antifungal agents. Bioorg Med Chem Lett 2008;18:141–6
- Emami S, Falahati M, Banifatemi A, Shafiee A. Stereoselective synthesis and antifungal activity of (Z)-trans-3-azolyl-2-methylchromanone oxime ethers. Bioorg Med Chem 2004;12:5881–9
- National Committee for Clinical Laboratory Standards. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard M27-A2; 2006; Wayne, Pennsylvania, USA
- Reulecke I, Lange G, Albrecht J, et al. Towards an integrated description of hydrogen bonding and dehydration: decreasing false positives in virtual screening with the HYDE scoring function. ChemMedChem 2008;3:885–97
- Schneider N, Hindle S, Lange G, et al. Substantial improvements in large-scale redocking and screening using the novel HYDE scoring function. J Comput Aided Mol Des 2012;26:701–23
- Emami S, Shafiee A. Stereoselective syntheses of (E)- and (Z)-2,3-dihydro-3-(1,2,4-triazolyl)-4H-1-benzopyran-4-one oxime ethers. Heterocycles 2001;55:2059–74
- Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol 1996;261:470–89