
Abstract
Acetylcholinesterase (AChE) inhibitors are yet the best drugs currently available for the management of Alzheimer’s disease. The recent phytochemical investigation has led to the isolation of a new depsidone 1 with moderate AChE activity (1 μg). This work was focused on its electronic properties analysed using commercially available programs. Both the active depsidone molecule 1 and galanthamine showed to have higher HOMO energies than the inactive depsidones 2–4, isolated from the same lichen species. However, the amino depsidone derivative 7, whose structure was proposed using computational approaches, is expected to be more active AChE inhibitor than the depsidone 1, due to the improved HOMO energy value. In addition, the molecular docking study indicated that the compound 7 has ability to make the well-known interactions of potent AChE inhibitors with the enzyme active site. The data presented herein support the design of novel AChE inhibitors based on the depsidone scaffold.
Introduction
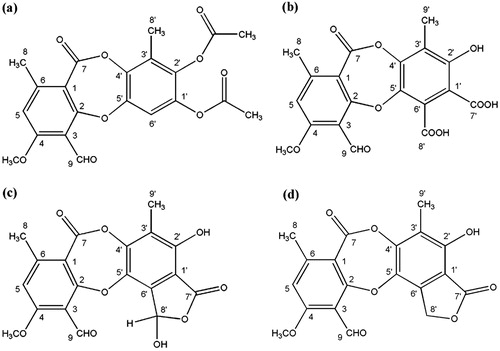
Acetylcholinesterase (AChE) inhibitors are yet the best drugs currently available for the management of Alzheimer’s diseaseCitation1. Because most of these inhibitors are alkaloids that often possess several side effects, such as galanthamine (GAL) used clinically for the disease treatmentCitation2, it is important to search for the novel inhibitors not belonging to this structural class. The recent phytochemical investigation conducted on a foliose lichen, Lobaria pulmonaria (L.) Hoffm. (Lobariaceae), has led to the isolation of a new depsidone in the form of diacetate derivative (1, ) with moderateCitation3 AChE inhibitory activity (1 μg). This is indeed the first and the only record of bioactive depsidone molecule in the search for the novel AChE inhibitors till date. The other new depsidone (2, ), stictic acid (3, ) and deoxystictic acid (4, ), all isolated from the same lichen speciesCitation3–5, showed no activity in the AChE inhibition testCitation6,Citation7. These experimental data have indicated the importance of covalent depsidone modification (acetylation) for the bioactivity observed. Although GAL inhibits the enzyme 100 times more than the compound 1 (0.01 µg), the depsidone scaffold is worthy of further studies due to the different chemical structure (11H-dibenzo[b,e][1,4]dioxepin-11-one ring system) from the alkaloid one. Therefore, the electronic properties of the bioactive depsidone 1 were investigated using commercially available programs. The selection of the computational method and basis set are crucial in getting accurate results. Zhao et al.Citation8,Citation9 have developed the M06 family of local and hybrid functionals, which, in most cases, have been found to be more accurate and have better performance than the most popular hybrid functional, B3LYP. The local functional M06-L applied in this study is reported to be very effective among the M06 familyCitation10. In order to understand the binding mode of the computationally proposed AChE inhibitor 7 based on depsidone scaffold, molecular docking simulation (a value added tool in medicinal chemistry) was performedCitation11–13.
Figure 1. (a) Depsidone 1; (b) Depsidone 2; (c) Stictic acid 3; and (d) Deoxystictic acid 4.
Material and methods
Calculation of the electronic properties
All calculations except drug likeness properties were performed with the Gaussian 09 suite of programsCitation14. M06-L functional of the M06 family was used. Since preliminary calculations showed that the studied systems had low ionisation potentials, diffuse functions were added to the heavy atoms. Stability of the wave function was found to be adequate under the perturbations considered. Furthermore, the geometry of all structures were fully optimised at M06-L level using the popular polarised basis set, 6-31 + G(d,p), which adds p functions to hydrogen atoms and d functions on heavy atoms. Frequency calculations were computed at the same level for each molecule to verify that the corresponding optimised geometries were real minimum without the imaginary frequency. Dipole moment (µ), HOMO-LUMO energies and related properties such as ionisation potential (I), electron affinity (A), electronegativity (χ), chemical hardness (η) and chemical softness (S) were calculated using optimised geometries. Drug likeness properties, i.e. octanol–water partition coefficient (ALogp), polar surface area (PSA) and number of hydrogen bond donors and accepters (HBD and HBA), were calculated using Accelrys Draw 4.1 software (Accelrys, Inc., San Diego, CA)Citation15.
Molecular docking study
Docking calculations were performed using the program AutoDock VinaCitation16. This software, an updated version of AutoDockCitation17, has better docking accuracy with a new scoring function as well as improved speed, efficient optimisation and multithreading properties.
AutoDockToolsCitation17 implemented in MGLTools (http://mgltools.scripps.edu/downloads) were used to prepare the inputs for docking. Protein data bank structure of AChE complexed with (–)-GAL at 2.3 Å resolution was downloaded from RCSB Protein Data Bank (1DX6). Binding site was determined using the GAL as the reference ligand. Hydrogens were added to the receptor, while all heteroatoms including the GAL were removed; default parameters were used. Discovery Studio Visualizer 4.0 (Accelrys, Inc., San Diego, CA)18 was used to analyse docking results and generate binding interactions between the ligand and the receptor.
Results and discussion
All the studied depsidone molecules have higher dipole moments than GAL but lower chemical hardness values, which is an indication of reactivity (). Sugimoto et al. have reported that AChE inhibitors with high dipole moments may have improved inhibitory activity towards the enzyme due to its remarkable large dipole momentCitation19.
Table 1. Electronic properties calculated at M06-L/6-31 + G(d,p) level.
Chemical hardness and softness are important electronic properties, which are a measure of the molecular stability and reactivity. Chemical hardness increases as the energy gap between HOMO and LUMO increases. A compound with low chemical hardness or high chemical softness can easily offer electrons to an acceptor. Nascimento et al. have indicated the importance of HOMO energy value for describing the interaction of AChE inhibitors with the receptorCitation20. Moreover, recent literature reveals that HOMO, LUMO and HOMO-1 make important contributions to the AChE inhibitory activityCitation21. The active depsidone 1 and GAL are found to have higher HOMO and HOMO-1 energies than the inactive depsidones 2–4. In fact, this trend is very clear for HOMO. In case of HOMO-1, the inactive depsidone 2 has higher HOMO-1 energy value than the active depsidone 1. It is noteworthy to mention that all the isolated depsidones, including the proposed ones, obey drug likeness properties except for the depsidone 2, which is yet known to be inactive. According to Veber et al., the depsidone 2 fails to obey the drug likeness property of having PSA being equal to or less than 140 Å2 (or having 12 or fewer H-bond donors and acceptors)Citation22.
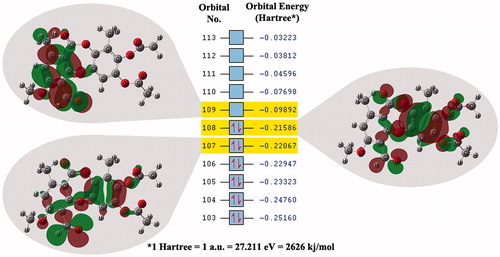
In the light of these results, the maps of frontier molecular orbitals for the depsidone 1 were calculated (). The arrangement of these frontier molecular orbitals is important to determine the most relevant part/atoms of the ligand for the interaction between the ligand and the receptor. It can be noticed that HOMO and HOMO-1 are mainly located on the ring with the acetate substituents.
Figure 2. Frontier molecular maps of the depsidone 1 calculated at M06-L/6-31 + G(d,p) level.
With the aim to design more potent AChE inhibitors based on the depsidone scaffold (following the fact that the active inhibitors have high HOMO and HOMO-1 energy values), the acetate groups were replaced with strong e-donating groups, i.e. –OH (5), –OCH3 (6) and –NH2 (7). After the optimisation of these newly proposed depsidone structures in the same way as the previous ones, their electronic properties were investigated. All three depsidone compounds were found to have both higher HOMO and HOMO-1 values than the reference molecule, the depsidone 1. In addition, the depsidone with –NH2 substituent (7) has even higher HOMO energy value than GAL itself ().
The molecular electrostatic potential maps of the depsidones 1 and 7 show potential regions of interaction with the AChE enzyme (). As it can be seen, high electron density lies on seven-membered ring, whereas positive region is located around the aromatic ring where acetylation occurs. The high electron densities (red colour) may act as proton acceptor region of the ligand. Actually, this high negative density has been reported to be able to transfer charges resulting in a π–π bonding interaction with the aromatic systems of the AChE active siteCitation19. Molecular docking simulation of depsidone 7 demonstrates that it makes π–π interactions with Trp 84, Phe 330 and Tyr 334 ().
Figure 3. Molecular electrostatic potential (top) and counter (bottom) maps of the depsidone 1 (a) and depsidone 7 (b), calculated at M06-L/6-31 + G(d,p) level.
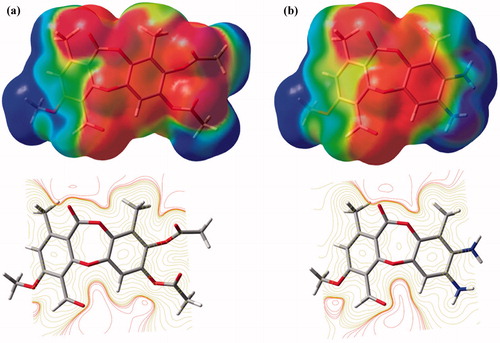
Figure 4. Binding interactions of the depsidone 7 with the AChE active site, based on molecular docking studies.
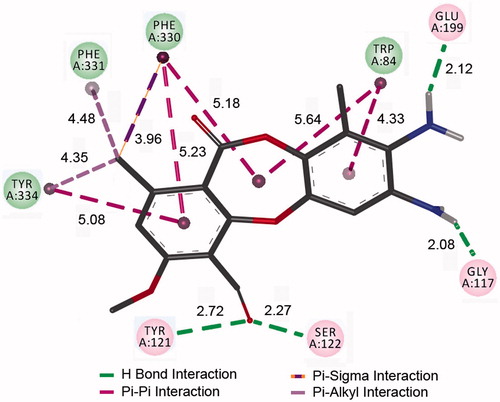
Campiani et al. have reported that the naturally occurring AChE inhibitor huperzine A makes a hydrogen bond with Glu 199 via a water bridge; if this water molecule could be incorporated into the inhibitor, the binding affinity might be improvedCitation23. They synthesised inhibitors that make hydrogen bond directly to Glu 199 improved the binding affinity. Molecular docking results clearly show that the depsidone 7 makes the same hydrogen bond with Glu 199, without the need for a water bridge (). This compound also establishes a π–σ interaction with Phe 330; π-alkyl interactions with Tyr 334 and Phe 331; hydrogen bonds with Gly 117, Tyr 121 and Ser 122. All these interactions are known to be formed between the potent AChE inhibitors and the enzyme active siteCitation24–26. Consequently, it may be supposed that the depsidone 7 does represent a promising lead for the new AChE inhibitors based on the depsidone scaffold.
Conclusions
According to the results obtained, the depsidone scaffold should be seriously considered in design of the novel AChE inhibitors. In such a search, the electronic property and molecular docking calculations could be used in the early stages that potentially save both money and time. Indeed, the substituents that positively affect e-donating ability might contribute to discover a new depsidone lead with an enhanced AChE inhibitory activity.
Declaration of interest
The authors report no declarations of interest.
References
- Graham LP. An introduction to medicinal chemistry. Oxford: University Press; 2005
- Scott LJ, Goa KL. Galantamine: a review of its use in Alzheimer’s disease. Drugs 2000;60:1095–122
- Pejin B, Tommonaro G, Iodice C, et al. A new depsidone of Lobaria pulmonaria with acetylcholinesterase inhibition activity. J Enzyme Inhib Med Chem 2013;28:876–8
- Pejin B, Tommonaro G, Iodice C, et al. A new lichen depsidone from Lobaria pulmonaria. Dig J Nanomater Bios 2012;7:1663–6
- Pejin B, Iodice C, Stanimirovic B, et al. A novel β-orcinol depsidone of the lichen Lobaria pulmonaria. Asian J Chem. [Epub ahead of print]
- Marston A, Kissling J, Hostettmann K. A rapid TLC bioautographic method for the detection of acetylcholinesterase and butyrylcholinesterase inhibitors in plants. Phytochem Anal 2002;13:51–4
- Rhee IK, van de Meent M, Ingkaninan K, Verpoorte R. Screening for acetylcholinesterase inhibitors from amaryllidaceae using silica gel thin-layer chromatography in combination with bioactivity staining. J Chromatogr A 2001;915:217–23
- Zhao Y, Truhlar DG. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J Chem Phys 2006;125:1–17
- Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc 2008;120:215–41
- Jacquemin D, Perpète EA, Ciofini I, et al. On the performances of the M06 family of density functionals for electronic excitation energies. J Chem Theory Comput 2010;6:2071–85
- Sakkiah S, Thangapandian S, John S, et al. 3D QSAR pharmacophore based virtual screening and molecular docking for identification of potential HSP90 inhibitors. Eur J Med Chem 2010;45:2132–40
- Ece A, Sevin F. The discovery of potential cyclin A/CDK2 inhibitors: a combination of 3D QSAR pharmacophore modeling, virtual screening, and molecular docking studies. Med Chem Res 2013;22:5832–43
- Ece A, Sevin F. Exploring QSAR on 4-cyclohexylmethoxypyrimidines as antitumor agents for their inhibitory activity of cdk2. Lett Drug Des Discov 2010;7:625–31
- Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision C.01. Wallingford (CT): Gaussian, Inc.; 2009
- Accelrys Software Inc. Accelrys Draw, Release 4.1. San Diego: Accelrys Software Inc.; 2013
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 2010;31:455–61
- Morris GM, Huey R, Lindstrom W, et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comput Chem 2009;16:2785–91
- Accelrys Software Inc. Discovery Studio Visualizer, Release 4.0. San Diego: Accelrys Software Inc.; 2013
- Sugimoto H, Sugimoto H, Yamanishi Y, et al. Donepezil hydrochloride (E2020) and other acetylcholinesterase inhibitors. Curr Med Chem 2000;7:303–39
- Nascimento ÉCM, Martins JBL, dos Santos ML, Gargano R. Theoretical study of classical acetylcholinesterase inhibitors. Chem Phys Lett 2008;458:285–9
- Nascimento ÉCM, Martins JBL. Electronic structure and PCA analysis of covalent and non-covalent acetylcholinesterase inhibitors. J Mol Model 2011;17:1371–9
- Veber DF, Johnson SR, Cheng H-Y, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002;45:2615–23
- Campiani G, Kozikowski AP, Wang S, et al. Synthesis and anticholinesterase activity of huperzine A analogues containing phenol and catechol replacements for the pyridone ring. Bioorg Med Chem Lett 1998;8:1413–18
- Contreras JM, Parrot I, Sippl W, et al. Design, synthesis, and structure-activity relationships of a series of 3-[2-(1-benzylpiperidin-4-yl)ethylamino]pyridazine derivatives as acetylcholinesterase inhibitors. J Med Chem 2001;44:2707–18
- Sippl W, Contreras JM, Parrot I, et al. Structure-based 3D QSAR and design of novel acetylcholinesterase inhibitors. J Comput Aided Mol Des 2001;15:395–410
- Carlier PR, Du D-M, Han Y-F, et al. Dimerization of an inactive fragment of huperzine A produces a drug with twice the potency of the natural product. Angew Chem Int Ed 2000;39:1775–7