
Abstract
This study reports on the design, synthesis and antiparasitic activity of three new semi-synthetic naphthoquinones structurally related to the naturally-occurring lapachol and lapachone. Of the compounds tested, 3-(3-methylbut-1-en-1-yl)-1,4-dioxo-1,4-dihydronaphthalen-2-yl acetate (1) was the most active against Plasmodium falciparum among both natural and semi-synthetic naphthoquinones, showing potent and selective activity. Compound 1 was able to reduce the in vitro parasite burden, in vitro parasite cell cycle, as well as the blood parasitemia in Plasmodium berghei-infected mice. More importantly, infection reduction under compound 1-treatment was achieved without exhibiting mouse genotoxicity. Regarding the molecular mechanism of action, this compound inhibited the hemozoin crystal formation in P. falciparum treated cells, and this was further confirmed by observing that it inhibits the β-hematin polymerization process similarly to chloroquine. Interestingly, this compound did not affect either mitochondria structure or cause DNA fragmentation in parasite treated cells. In conclusion, we identified a semi-synthetic antimalarial naphthoquinone closely related to isolapachol, which had stronger antimalarial activity than lapachol.
Introduction
Malaria represents a major threat to the public health worldwide, with over 250 million clinical cases and two million deaths annuallyCitation1,Citation2. Most Plasmodium falciparum isolates in endemic areas have developed multi-resistance to the aminoquinolines (chloroquine, amodiaquine and mefloquine)Citation3. Because of this, artemisinin derivatives are recommended in combination with aminoquinolines and other drugs to treat the infection caused by multidrug resistant strainsCitation4. Resistance to artemisinin has been reportedCitation5, making the development of new antimalarial drugs necessary.
Naphthoquinones are a useful chemical class for antiparasitic drug developmentCitation6–10. This is well exemplified by buparvaquone, parvaquone and atovaquone, the latter being used in a fixed-dose combination with proguanil for treating drug-resistant P. falciparum malariaCitation11–13. Based on this, other naphthoquinones have been investigated as antimalarials in potential. Specifically, the naturally-occurring naphthoquinones lapachol and α- and β-lapachones have a broad spectrum antimalarial activity against the erythrocytic and liver stages in both chloroquine-sensitive and chloroquine-resistant strainsCitation6,Citation14. Like atovaquone, these naphthoquinones affect Plasmodium growth, in part, by impairing mitochondrial function, which affects the electron transport flux and subsequently prevents hemozoin formationCitation15,Citation16. In view of this, naphthoquinones are considered promising antimalarial agents. In recent years, chemical modifications in the naphthoquinone backbone, as well as in the side chain, have identified derivatives with enhanced antimalarial activityCitation17–22.
Based on the structure–activity relationship observed for lapachol and α- and β-lapachones, here we prepared for the first time three derivatives denoted as compounds 1–3 and evaluated the antimalarial activity. Compound 1 is the acetylated form of isolapachol, while compounds 2 and 3 are semi-synthetic naphthopyrandiones containing an iodine substituent. By evaluating the antiparasitic activity of these derivatives and comparing the potency to their naturally-occurring analogs, we observed an enhancement of activity for compound 1 in comparison to lapachol. In addition, compound 1 was able to inhibit the Plasmodium life cycle in erythrocytes, inhibited hematin crystal formation, but had little effect on Plasmodium mitochondrial activity. Finally, compound 1 was able to reduce blood parasitemia in P. berghei-infected mice, and importantly, this was achieved without exhibiting mouse toxicity or genotoxicity.
Materials and methods
Compound preparation
2-Hydroxy-3-(3-methylbut-2-en-1-yl)naphthalene-1,4-dione (lapachol) was extracted from the bark of Tabebuia aurea (Manso) S. Moore by using a previously described methodCitation23. Yellow crystals of compound 1 were obtained in a 1–2 % yield from the dry bark. M.p. 137–139 °C (literature 140 °C). 2-Hydroxy-3-(3-methylbut-1-en-1-yl)naphthalene-1,4-dione (isolapachol) was prepared from lapachol according to the methodology described in reference 25. Yellow crystals, m.p. 76–77 °C. 2,2-Dimethyl-3,4-dihydro-2H-benzo[g]chromene-5,10-dione (α-lapachone) was prepared from lapachol following a modified Hooker procedureCitation24. Red crystals, m.p. 152–154 °C. Yield = 76%. 2,2-Dimethyl-3,4-dihydro-2H-benzo[h]chromene-5,6-dione (β-lapachone) was prepared from lapachol following the methodology described above. Yellow crystals, m.p. 113–115 °C. Yield = 14 % ().
Figure 1. Structures of naphthoquinones investigated here as anti-P. falciparum compounds.
3-(3-Methylbut-1-en-1-yl)-1,4-dioxo-1,4-dihydronaphthalen-2-yl acetate (1) in a 100 mL round bottom flask, isolapachol (2.0 g, 8.2 mmol) was dissolved in 13 mL of acetic anhydride and stirred at room temperature. Anhydrous NaOAc (2.0 g, 3.9 mmol) was added to the mixture and the reaction mixture was then maintained under stirring for 30 min, under heating at 100 °C. After this time, the mixture was cooled to room temperature, ice was added, which was followed by compound precipitation. The solid was recrystallized in ethanol. Yellow crystals, m.p. 75.6–76.3 °C, Yield = 54%. 1H NMR (200 MHz, CDCl3, δ): 1.09 (3H, s, CH3), 1.13 (3H, s, CH3), 2.42 (3H, s, –OCOCH3), 2.47–2.60 (1H, m, H-3′), 6.36 (1H, d, J 16.2 Hz, H-1′), 6.95 (1H, dd, J = 7.2, 16.2 Hz, H-2′), 7.68–7.75 (2H, m, H-6 and H-7), 8.05–8.13 (2H, m, H-5 and H-8). HRESIMS calculated for C17H16O4: 284.1048; found: 285.1079 [M + 1]+.
3-Iodo-2,2-dimethyl-3,4-dihydro-2H-benzo[g]chromene-5,10-dione (2) was prepared from α-lapachone following the methodology previously describedCitation25. Yellow crystals, m.p. 108–112 °C. Yield = 36%. HRESIMS calculated for C15H13O3I: 367.9909; found: 368.9965 [M + 1]+.
3-Iodo-2,2-dimethyl-3,4-dihydro-2H-benzo[h]chromene-5,6-dione (3) was prepared from β-lapachone following the previously described methodCitation25. Red crystals, m.p. 137–138 °C. Yield = 54%. HRESIMS calculated for C15H13O3I: 367.9909; found: 369.0011 [M + 1]+.
Mice and cells
BALB/c and Swiss Webster mice were supplied by the animal breeding facility at Centro de Pesquisas Gonçalo Moniz (Salvador, Brazil) and maintained in sterilized cages under a controlled environment, receiving a balanced diet for rodents and water ad libitum. Spleens of BALB/c mice were removed in accordance with the guidelines of the Commission for Experiments in Laboratory Animals. P. falciparum (W2 strain) was maintained in a continuous culture of human erythrocytes (blood group O+) using Roswell Park Memorial Institute-1640 (RPMI-1640) medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% human plasma. BALB/c mouse splenocytes were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal calf serum (Cultilab, Campinas, Brazil) and 50 μg/mL of gentamycin (Novafarma, Anapolis, Brazil). Mefloquine and chloroquine were kindly supplied by FarManguinhos (Rio de Janeiro, Brazil), and cyclophosphamide was purchased from Sigma-Aldrich. Drugs were dissolved in DMSO (Sigma-Aldrich) and diluted in media to a final concentration of 0.5% dimethylsulfoxide (DMSO). All procedures described here had prior approval from the local animal ethics committee.
Cytotoxicity in splenocytes
BALB/c mouse splenocytes (6 × 105 cells/well) were distributed in a 96-well plate. Compounds previously dissolved in DMSO were diluted in DMEM and added to the cell cultures. Five compound concentrations were used, each one in triplicate. Cells were incubated with drugs for 24 h, then 1.0 µCi/well [3H]-thymidine (PerkinElmer, Boston, MA) was added to the cultures and incubated for 24 h at 37 °C and 5% CO2. After this time, cells were harvested using the cell harvester (Brandel MPXRI 96TI, Gaithersburg, MD) and the [3H]-thymidine incorporation was determined by using a β-radiation counter (Hidex Multilabel Reader, Turku, Finland). Cell viability was determined in comparison to untreated cultures. The cytotoxic concentration to 50% (CC50) was calculated, using non-linear regression. Two independent experiments in triplicate were performed.
In vitro anti-P. falciparum activity
Parasites grown at 1–1.5% parasitemia and 2.5% hematocrit were distributed into 96-wells plate. Compounds were dissolved in DMSO followed by serial dilution in RPMI-1640 medium without hypoxanthine. Five compound concentrations were used, each one in triplicate. After 24 h incubation time, 0.5 µCi/well [3H]-hypoxanthine monochloride (PerkinElmer, Boston, MA) was added, incubated for 24 h and then cells were harvested using the cell harvester (Brandel MPXRI 96TI) and the [3H]-hypoxanthine incorporation was determined by using a β-radiation counter (Hidex Multilabel Reader). Inhibition of parasite growth was determined in comparison to untreated cultures. The inhibitory concentration to 50% (IC50) was calculated using non-linear regression. Two independent experiments in triplicate were performed.
Activity on the P. falciparum cell cycle
Parasites grown at 2% parasitemia and 2.5% hematocrit were synchronized to ring stage by using sorbitol (5%). Cells were then treated with compound 1 and incubated until 48 h at 37 °C. Thin blood films were prepared and stained with fast panoptic (Laborclin, Pinhais, Brazil) and examined by optical microscopy. Parasitemia was determined by counting at least 1000 erythrocytes per multiple sections of each slide and the results were pooled and data analyzed for statistical significance. Two independent experiments in triplicate were performed.
Transmission electron microscopy (TEM)
Parasite cultures were treated with 2.3 µM of compound 1 and incubated for 24 h at 37 °C. Untreated parasites were used as controls. After incubation, the parasites were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) and post-fixed with the same buffer containing 1% OsO4, 0.8% potassium ferricyanide, and 5.0 mM calcium chloride for 40 min at room temperature, in the absence of light. Then, parasites were washed in the same buffer and dehydrated on increasing concentrations of acetone (30–100%) for 10 min in each concentration. Polymerization was achieved with polybed epoxy resin (Polysciences, Warrington, FL). Ultrathin slices were contrasted with uranyl acetate and lead citrate and observed in an EM109 electron microscope (Zeiss, Oberkochen, Germany).
Hemolysis assay
Fresh human erythrocytes (type O+) were washed three times in PBS and resuspended in the same buffer to a 1% hematocrit solution, from which 100 µL were distributed per well in 96-well plates. Compound 1, previously dissolved in phosphate buffered saline, was added at 200 µM, in triplicate, and plates were incubated for 1 h at 37 °C. Saponin (Sigma-Aldrich) was used as a hemolytic drug at 1% v/v. After incubation, plates were centrifuged (1500 rpm for 10 min) and 100 μL of each supernatant was transferred to a microtiter plate. Released hemoglobin was quantified by measuring the absorbance at 540 nm in a spectrophotometer. The percentage of hemolysis was calculated in comparison to untreated cells. Two independent experiments in triplicate were performed.
Inhibition of β-hematin formation
Fifty microliters of a freshly prepared solution of hemin (0.5 mg/mL) dissolved in 0.2 M NaOH were mixed to a 75 μL of 3 M sodium acetate, 25 μL of 17.4 M acetic acid and 50 μL of the compounds. Each compound was tested in five concentrations, each one in triplicate. After 24 h of incubation at 37 °C, the resulting solution was spun down for 15 min at 3500 rpm and the pellet was washed with 200 μL of DMSO. This step was repeated once and, after a final wash with water, the pellet was dissolved in 0.1 M NaOH (150 μL). The absorption was read at 405 nm using a spectrophotometer (SpectraMAX model 190, Molecular Devices, Sunnyvale, CA). Results are expressed as percentage of inhibition of hematin formation in comparison to the negative control (without the compounds). Experiments were carried out at least twice, using compound concentrations in triplicate.
Determination of P. falciparum DNA integrity by TUNEL assay
DNA fragmentation was tested through a TUNEL assay kit (APO-DIRECT™, BD Pharmingen, San Jose, CA). P. falciparum ring stages were cultured with the compound 1 (2.3, 11.7 and 23.5 μM) for 24 h at 37 °C. Free parasites were obtained from their host erythrocytes by treatment with 0.1 % saponin (w/v) in PBS. Untreated parasite (negative control) was included in the experiment. The parasites were washed with PBS (1500 rpm for 5 min), fixed for 1 h at 4 °C with 1% paraformaldehyde in PBS and permeabilized with 70% ice-cold ethanol according to the manufacturer's recommendations. Reaction solution was incubated for 1 h at 37 °C. The total DNA was stained by incubation with a solution containing propidium iodide (PI)/ Ribonuclease (RNase). Fluorescein isothiocyanate–deoxyuridine triphosphate (FITC-dUTP) was analyzed at 520 nm, while PI was at 623 nm. Deoxynuclease (DNase, Life Technologies, Foster City, CA) was used as positive control at 37 °C for 30 min at 2 units/µL. Cell acquisition was performed using a BD Calibur flow cytometer by acquiring 10 000 events and data were analyzed by BD CellQuest. Two independent experiments in triplicate were performed.
Determination of parasite mitochondrial membrane potential (ΔΨm)
Changes in parasite mitochondrial membrane potential were determined with 3,3′-dihexyloxacarbocyanine iodide probe (DiOC, Life Technologies). Parasitized red blood cells enriched for trophozoite stage were resuspended in RPMI medium and incubated with DiOC (2 nM final concentration) for 20 min at 37 °C. The compound (2.3, 11.7 and 23.5 μM) was then added and incubated for 20 min. Carbonyl cyanide m-chlorophenylhydrazone (CCCP, Sigma-Aldrich) was used as a positive control at 100 µM. Changes in the fluorescence by DiOC incorporation into mitochondria membranes of parasitized red blood cells were evaluated using a BD Calibur flow cytometer by acquiring 10 000 events, and data were analyzed by BD CellQuest software.
Antiparasitic activity in P. berghei-infected mice
Swiss Webster mice were infected by intraperitoneal route with 106 red blood cells infected with the P. berghei NK65 strain. Animals were randomly sorted into n = 5/group. Two hours post-infection, treatment was given once a day during four consecutive days. Drugs were dissolved in saline containing 20% DMSO and each mouse received 200 µL by intraperitoneal or oral (gavage) route. Untreated infected group received only saline containing 20% DMSO. The antimalarial activity was evaluated by counting parasitemia in blood smears in the 4–8 d post-infection. Cells were fixed in methanol, stained with fast panoptic (Laborclin, Pinhais, Brazil) and examined by optical microscopy. Mortality was followed up until 30 d post-infection.
Micronucleus test
Uninfected Swiss Webster mice (male, 20–25 g, n = 5) were treated by intraperitoneal injection with 100 mg/kg body weight of compound 1. Eighteen to 24 h after treatment, femurs from each animal were dissected and bone marrow was washed with fetal bovine serum (Gibco, Foster City, CA). Bone marrow smears were prepared on glass slides after centrifugation at 2000 rpm (5 min). Slides were air-dried and stained using Wright-Giemsa and evaluated using an optical microscope. The polychromatic erythrocyte (PCE) and mononucleated polychromatic erythrocyte (NPCE) number and ratio were established for each animal by scoring a total of 8000 erythrocytes (PCE + NPCE). Bone marrow preparations were evaluated for the presence of micronuclei by counting 1000 PCE for each animal. One experiment was performed.
Statistical analysis
Nonlinear regression was calculated in GraphPad Prism version 4.0 (Graph Pad software, San Diego, CA). Two way ANOVA test was used and differences were considered significant for p < 0.05.
Results
Antiparasitic activity and cytotoxicity
The antiparasitic activity was evaluated in intraerythrocytic forms of P. falciparum W2 strain. Cytotoxicity was evaluated in mouse splenocytes. Mefloquine was used as an antimalarial reference drug. Activity was described in term of IC50 and CC50 values (). Lapachol exhibited low antiparasitic activity, while isolapachol did not show activity (data not shown). Compound 1 was substantially more active than lapachol, displaying an IC50 of 2.3 ± 0.1 μM. α- and β-lapachones exhibited IC50 of 15.8 ± 3.5 and 20.5 ± 1.0 μM, respectively, being more potent than lapachol. The synthetic lapachone derivatives 2 and 3 displayed equal antiparasitic activity as observed for naturally-occurring lapachones. Following this, cytotoxicity to mouse splenocytes was analyzed. In comparison to mefloquine, which exhibited CC50 of 5.2 ± 0.4 μM, lapachol and compound 2 were less cytotoxic. Other compounds exhibited similar or higher cytotoxicity than mefloquine. Regarding the selectivity index, compound 1 presented higher selectivity than lapachol, although it was less selective than mefloquine.
Table 1. In vitro antiparasitic activity and cytotoxicity.
P. falciparum cell cycle
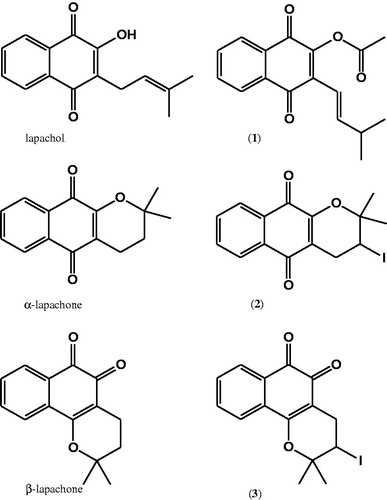
Compound 1 was the most potent antiparasitic naphthoquinone, and thus was selected to evaluate its effect in the parasite intracellular development in host cells. In this assay, W2 strain of P. falciparum was synchronized to ring-stage and incubated until 48 h in the presence of compound 1 at 2.3 μM (IC50) and 4.7 μM (IC100). The distribution of rings, schizonts and trophozoites in erythrocytes was counted by optical microscope. In untreated cells, ring-stage parasites maturate to trophozoites after 24 h of incubation and at 48 h of incubation, a number of schizonts are observed. As can be seen in , compound 1 at 2.3 µM substantially reduced the ring-stage maturation into schizonts. In addition, compound 1 treatment prevented the formation of schizonts in the cell culture. Furthermore, we evaluated the hemolytic effect of compound 1 in uninfected erythrocytes. As shown in , it did not induce hemolysis in concentrations up to 200 μM.
Figure 2. Antiparasitic activity of compound 1 in the cell-cycle development of blood-stage P. falciparum. Erythrocytes were infected with W2 strain P. falciparum (synchronized to ring stage) and incubated with compound 1 at 2.3 and 4.7 µM during one cycle. Parasites were analyzed by optical microscopy in different times. Significance in reference to % of parasitemia: *p < 0.05; ***p < 0.001.
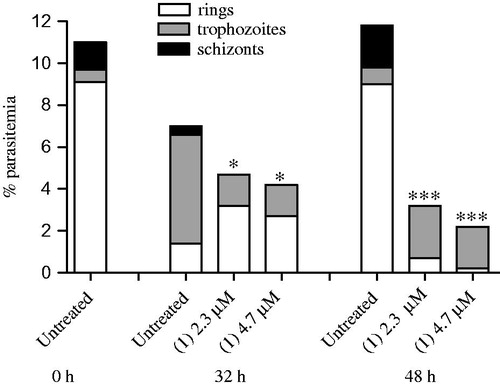
Table 2. Effects on the hematin formation and hemolysis.
Investigating the mechanism of action
We used TEM to examine the ultrastructural morphology of intraerythrocytic parasites treated with compound 1. In untreated parasite, the presence of needle-like structures characteristic of hemozoin formation is observed (). In contrast to the untreated control, compound 1 treatment inhibited the formation of hemozoin crystals inside of digestive parasitic vacuoles. Moreover, it was observed that compound 1 treatment alters morphology in parasite cells, characteristic of parasiticidal agents. To corroborate to the hemozoin inhibition finding, the ability of compound 1 to inhibit the β-hematin formation was determined. Chloroquine, the reference drug, inhibited β-hematin formation with an IC50 of 0.86 ± 0.30 mM. Compound 1 exhibited similar property of β-hematin inhibition, while lapachol exhibited low activity ().
Figure 3. TEM analysis of P. falciparum. Parasites were treated with 2.3 µM of compound 1 and incubated for 24 h. Panels A and B show untreated parasites, highlighting the presence of hemozoin crystals inside the digestive vacuoles. Panel C and D show treated parasites, where is possible to observe the hemozoin crystal absence. Panel E exhibits the number of parasites containing hemozoin crystals.
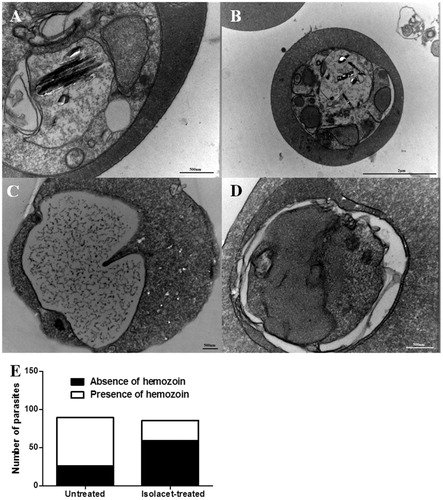
In the second set of experiments, we treated W2 strain P. falciparum with different concentrations of compound 1, incubated for 24 h and evaluated mitochondria impairment as well as DNA fragmentation. Cells were stained with DIOC or by using TUNEL kit assay and examined by flow cytometry. When compared to untreated cells, CCCP-treatment caused cell depolarization, leading to cells positively stained for DIOC. In contrast, parasite cells treated with compound 1 were not positively stained for DIOC (data not shown). Therefore, compound 1-based treatment does not cause parasite cell death through mitochondria impairment. By using TUNEL kit assay, an increase in the percentage of stained parasites was observed during DNase treatment at 2 units/mL in comparison to untreated parasites (). However, parasites treated with compound 1 at different concentrations did not present positively stained cells, indicating that this compound does not cause DNA fragmentation in parasitic cells.
Figure 4. DNA fragmentation in W2 strain P. falciparum incubated with compound 1. Parasites were incubated with 0.05% DMSO (untreated), DNAse (positive control) or compound 1 and then analyzed by flow cytometry by using to the terminal deoxynucleotidyltransferase-mediated dUTP nick end labelling assay (TUNEL). One experiment was performed, in triplicate. *p < 0.001 in comparison to untreated cells.
P. berghei-infected mice
Based on the results described above, we evaluated the activity of compound 1 in P. berghei-infected Swiss mice. Infected mice receiving saline (untreated group) showed high blood parasitemia after day 4 of parasite inoculation and peaked on day 6. Chloroquine given orally at 50 mg/kg once a day for 4 d starting from day 1 after infection reduced blood parasitemia (p < 0.001) (, ). Compared to untreated infected mice, intraperitoneal treatment with compound 1 at a dose of 10 mg/kg did not reduce parasitemia, while at doses of 50 and 100 mg/kg, a reduction of parasitemia peak was observed (p < 0.001). Compound 1 given orally at 200 mg/kg reduced 49% blood parasitemia at day 8 in comparison to untreated infected group, however, this was lower than observed under intraperitoneal treatment at a dose of 100 mg/kg. Mice mortality was monitored up to 30 d post-infection. In comparison to untreated group, chloroquine treatment decreased mortality (log rank, p < 0.001). The groups treated with 100 mg/kg compound 1 showed no significant difference in mortality rate to untreated group.
Figure 5. (A) Blood parasitemia and (B) survival in P. berghei-infected mice. Swiss Webster mice (n = 5/group) were infected with P. berghei and then treated daily for four consecutive days. Compound 1 was given by intraperitoneal injection, chloroquine by oral gavage. Values represent the means ± SEM of one experiment (n = 5). (A) *p < 0.05; **p < 0.01; ***p < 0.001 in comparison to vehicle group. (B) According to the Log-rank (Mantel Cox) analysis, there is no statistically significance between vehicle and compound 1.
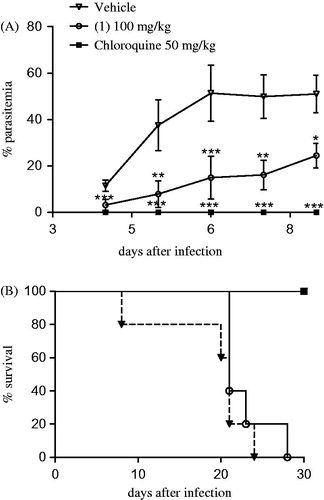
Table 3. Antimalarial activity in P. berghei-infected mice.
Micronucleus test
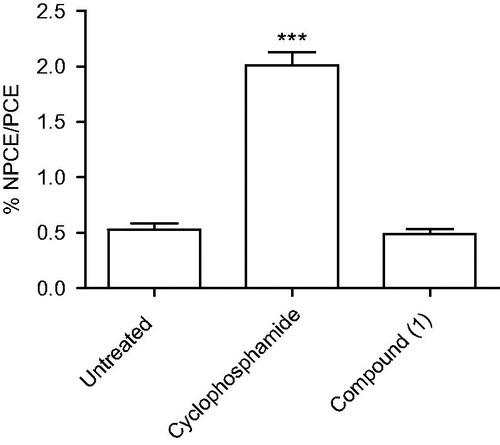
We evaluated the genotoxic activity of compound 1 in inducing the formation of micronucleus in PCEs. In a control experiment, Swiss Webster mice receiving saline (untreated group) showed a low frequency of micronucleated cells. The reference drug cyclophosphamide given intraperitoneally at 50 mg/kg increased the frequency of micronucleated cells in comparison to untreated group (p < 0.001) (). In contrast, intraperitoneal treatment of compound 1 at dose 100 mg/kg did not increase the frequency of micronucleated cells in comparison to the untreated group.
Figure 6. Frequency of micronucleated polychromatic erythrocytes (NPCE) observed in Swiss Webster mouse bone marrow. Swiss mice (n = 5/group) received intraperitoneal injection of saline (untreated), cyclosphophamide (50 mg/kg) and compound 1 (100 mg/kg). After 18 to 24 h of treatment, the percentage of NPCE was determined relatively to 8000 polychromatic erythrocytes (PCE) per group. Values represent the means ± SEM of one experiment (n = 5/group). ANOVA, ***p < 0.001 in comparison to untreated group.
Discussion
An effective antimalarial treatment continues to be necessary due to acquired resistance of P. falciparum to aminoquinoline and artemisin regimes. Screening of isolated natural and semi-synthetic compounds is a reliable strategy to identify new anti-P. falciparum agents. Naphthoquinones are a rich source of antimalarial compounds. Of these, lapachol and lapachones are some of the most investigated in terms of potency and selectivity. In the present study, we determined the anti-P. falciparum activity of acetylated isolapachol denoted here as compound 1 and of two semi-synthetic naphthopyrandiones (2 and 3) that features an iodine substituent attached in the quinone backbone.
We observed that naphthoquinones (1–3) are anti-Plasmodium agents. Despite the gain in lipophilicity and bulky characteristics usually observed upon halogen atom attachment in drugsCitation26, here the incorporation of an iodine substituent into α-lapachone (2) and β-lapachone (3) did not improve the antimalarial activity. In contrast, we observed a substantial enhancement of activity for compound 1 in comparison to lapachol. More specifically, it was observed that compound 1 strongly inhibits the erythrocytic form of W2 strain P. falciparum at nontoxic concentrations for mouse macrophages. In addition, compound 1 is able to prevent the parasite development into schizontsCitation27 and this was achieved without causing hemolysis.
An examination of parasite morphology revealed that compound 1 inhibits the formation of hemozoin crystals inside the digestive vacuoles. In agreement to this, it was observed that compound 1 but not lapachol, inhibits the β-hematin formation and this may explain the enhanced antimalarial activity of compound 1 versus lapachol. Hemozoin is vital for parasite maintenanceCitation28. In fact, we observed that most parasite cells were destroyed under compound 1 treatment, suggesting this is a parasiticidal agent. Flow cytometry analysis revealed that compound 1 did not change the mitochondrial transmembrane potential as well as did not cause DNA fragmentation in P. falciparum cell culture.
Most importantly, we analyzed compound 1 antimalarial activity effects in the in vivo infection. This naphthoquinone was able to reduce the blood parasitemia in a dose-dependent manner. On the one hand, the highest parasitemia reduction was observed under a dose of 100 mg/kg by intraperitoneal injection. As limitation, this compound did not show substantial antimalarial activity when given orally, probably because it is susceptible to gastric hydrolysis. As advantage, this compound did not exhibit in vivo genotoxicity when evaluated at the same dose it exerts optimal antimalarial activity. Altogether, these findings reinforce that naphthoquinones are active antimalarial agents and at the same time do not cause hemolysis and genotoxicity. Therefore, the screening for new anti-P. falciparum naphthoquinones is an attractive line of drug development.
Conclusion
Three new naphthoquinones were investigated as antimalarial compounds. Of these, the acetylated isolapachol, denoted here as compound 1, exhibited the highest potency and selectivity. This compound inhibits the parasite growth as well as the intracellular development, behaves as a parasiticidal agent and, in parts, exerts antiparasitic activity through inhibiting the formation of hemozoin crystals. Importantly, this compound reduces blood parasitemia in P. berghei-infected mice without causing hemolysis or genotoxicity, therefore, suggesting this compound as a starting point for drug design and development of antimalarial agents.
Declaration of interest
The authors have declared that no competing interests exist.
This work received fundings from Conselho Nacional de Pesquisas Brasileira (CNPq), Fundação de Amparo as Pesquisas do Estado da Bahia (FAPESB), Fundação de Amparo à Pesquisa do Estado de Alagoas (FAPEAL) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). M.B.P.S., M.O.G.S., A.E.G.S., C.A.C., J.M.B.F. and R.R.S hold a CNPq fellowship. T.S.M. is holding a FAPESB doctoral scholarship, while D.R.M.M. is holding a FAPESB scholarship for visiting scholars.
References
- Cotter C, Sturrock HJ, Hsiang MS, et al. The changing epidemiology of malaria elimination: new strategies for new challenges. Lancet 2013;382:900–11
- William T, Rahman HA, Jelip J, et al. Increasing incidence of Plasmodium knowlesi malaria following control of P. falciparum and P. vivax Malaria in Sabah, Malaysia. PLoS Negl Trop Dis 2013;7:e2026
- Lewis IA, Wacker M, Olszewski KL, et al. Metabolic QTL analysis links chloroquine resistance in Plasmodium falciparum to impaired hemoglobin catabolism. PLoS Genet 2014;10:e1004085
- Baird JK. Effectiveness of antimalarial drugs. N Engl J Med 2005;352:1565–77
- Dondorp AM, Nosten F, Yi P, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 2009;361:455–67
- Carvalho LH, Rocha EM, Raslan DS, et al. U. In vitro activity of natural and synthetic naphthoquinones against erythrocytic stages of Plasmodium falciparum. Braz J Med Biol Res 1988;21:485–7
- Hudson AT, Randall AW, Fry M, et al. Novel anti-malarial hydroxynaphthoquinones with potent broad spectrum anti-protozoal activity. Parasitology 1985;90:45–55
- Pinto AV, Ferreira VF, Capella RS, et al. Activity of some naphthoquinones on blood stream forms of Trypanosoma cruzi. Trans Royal Soc Trop Med. Hyg 1987;81:609–10
- Santhamma KR and Raj RK. Quinone analogues: a drug of choice for the control of filariasis. Biochem Biophys Res Commun 1993;190:201–6
- Ferguson DJ, Huskinson-Mark J, Araujo FG, Remington JS. An ultrastructural study of the effect of treatment with atovaquone in brains of mice chronically infected with the ME49 strain of Toxoplasma gondii. Int J Exp Pathol 1994;75:111–16
- Beerahee M. Clinical pharmacology of atovaquone and proguanil hydrochloride. J Travel Med 1999;6:S13–17
- Baggish AL, Hill DR. Antiparasitic agent atovaquone. Antimicrob Agents Chemother 2002;46:1163–73
- Nixon GL, Moss DM, Shone AE, et al. Antimalarial pharmacology and therapeutics of atovaquone. J Antimicrob Chemother 2013;68:977–85
- Pérez-Sacau E, Estévez-Braun A, Ravelo AG, et al. Antiplasmodial activity of naphthoquinones related to lapachol and beta-lapachone. Chem Biodiversity 2005;2:264–74
- Henry M, Alibert S, Rogier C, et al. Inhibition of efflux of quinolines as new therapeutic strategy in malaria. Curr Top Med Chem 2008;8:563–78
- Hughes LM, Lanteri CA, O'Neil MT, et al. Design of anti-parasitic and anti-fungal hydroxy-naphthoquinones that are less susceptible to drug resistance. Mol Biochem Parasitol 2011;177:12–19
- De-Andrade-Neto VF, Goulart MO, da Silva-Filho JF, et al. Antimalarial activity of phenazines from lapachol, beta-lapachone and its derivatives against Plasmodium falciparum in vitro and Plasmodium berghei in vivo. Bioorg Med Chem Lett 2004;14:1145–9
- Lanfranchi DA, Cesar-Rodo E, Bertrand B, et al. Synthesis and biological evaluation of 1,4-naphthoquinones and quinoline-5,8-diones as antimalarial and schistosomicidal agents. Org Biomol Chem 2012;10:6375–87
- Hussain H, Specht S, Sarite SR, et al. New quinoline-5,8-dione and hydroxynaphthoquinone derivatives inhibit a chloroquine resistant Plasmodium falciparum strain. Eur J Med Chem 2012;54:936–42
- Schuck DC, Ferreira SB, Cruz LN, et al. Biological evaluation of hydroxynaphthoquinones as anti-malarials. Malaria J 2013;12:234. doi:10.1186/1475-2875-12-234
- Sharma A, Santos IO, Gaur P, et al. Addition of thiols to o-quinone methide: new 2-hydroxy-3-phenylsulfanylmethyl [1,4]naphthoquinones and their activity against the human malaria parasite Plasmodium falciparum (3D7). Eur J Med Chem 2013;59:48–53
- De-Rezende LC, Fumagalli F, Bortolin MS, et al. In vivo antimalarial activity of novel 2-hydroxy-3-anilino-1,4-naphthoquinones obtained by epoxide ring-opening reaction. Bioorg Med Chem Lett 2013;23:4583–6
- Barbosa-Filho JM, Lima CSA, Amorim ELC, et al. Botanical study, phytochemistry and antimicrobial activity of Tabebuia aurea. Int J Exp Bot 2004;73:221–8
- Barbosa TP, Camara CA, Silva TMS, et al. New 1,2,3,4-tetrahydro-1-aza-anthraquinones and 2-aminoalkyl compounds from norlapachol with molluscicidal activity. Bioorg Med Chem 2005;13:6464–9
- Barbosa-Filho JM, Silva TMS, Giulietti AM, et al. Brazilian patent, PI 0403686-7; 2006, 12 pages. CAN 147:448582
- Hernandes MZ, Cavalcanti SM, Moreira DR, et al. Halogen atoms in the modern medicinal chemistry: hints for the drug design. Curr Drug Targets 2010;11:303–14
- Chalapareddy S, Bhattacharyya MK, Mishra S, Bhattacharyya S. Radicicol confers mid-schizont arrest by inhibiting mitochondrial replication in Plasmodium falciparum. Antimicrob Agents Chemother 2014;58:4341–52
- Coronado LM, Nadovich CT, Spadafora C. Malarial hemozoin: from target to tool. Biochim Biophys Acta 2014;1840:2032–41