
Abstract
Transthyretin (TTR) is a 54 kDa homotetrameric protein that transports thyroxine (T4) and retinol (vitamin A), through its association with retinol binding protein (RBP). Under unknown conditions, it aggregates to form fibrils associated with TTR amyloidosis. Ligands able to inhibit fibril formation have been studied by X-ray crystallography. The use of polyethylene glycol (PEG) instead of ammonium sulphate or citrate has been evaluated as an alternative to obtain new TTR complexes with (R)-3-(9-fluoren-9-ylideneaminooxy)-2-methyl-N-(methylsulfonyl) propionamide (48R(1)) and 2-(9H-fluoren-9-ylideneaminooxy) acetic acid (ES8(2)). The previously described fluorenyl based inhibitors (S)-3-((9H-fluoren-9-ylideneamino)oxy)-2-methylpropanoic acid (6BD) and 3-((9H-fluoren-9-ylideneamino)oxy)propanoic acid (7BD) have been re-evaluated with the changed crystallization method. The new TTR complexes with compounds of the same family show that the 9-fluorenyl motif can occupy alternative hydrophobic binding sites. This augments the potential use of this scaffold to yield a large variety of differently substituted mono-aryl compounds able to inhibit TTR fibril formation.
Introduction
Transthyretin (TTR) is a stable protein composed of four identical subunits of 14 KDa each. The acronym TTR denotes the protein’s functions: transporter, thyroxine, and retinol. TTR is synthesized mainly by the liver and the choroid plexus of the brainCitation1 and in minor amounts in the retinaCitation2 and in human placentaCitation3. It is present both in human plasma and in the cerebrospinal fluid (CSF) at different concentrationsCitation4. The TTR tetramer presents two identical funnel-shaped T4 binding sites, located at a dimer–dimer interface and orthogonal to the RBP–vitamin A complex sites. The T4 binding site has a small inner and a larger outer binding subsites. Furthermore, in the active site three pairs of symmetric hydrophobic depressions are present, which are occupied by the iodine atoms of the natural ligand (T4), and are ascribed to as the halogen binding pockets (HBP1, HBP2, and HBP3)Citation5.
TTR normally circulates as a soluble protein, but in some TTR mutation carriers or in elderly with WT-TTR it forms, with an unknown molecular mechanism, amyloid fibrils that are responsible of TTR amyloidosis (http://amyloidosismutations.com).
These pathologies are characterized by systemic neuropathies and cardiomyopathies that lead, progressively, to severe life-threatening conditionsCitation6–10. An accepted strategy to treat the hereditary ATTR is the transplantation of amyloidogenic TTR-producing liver with one that produces only wild type TTR, with the aim to decrease the circulating concentration of unstable TTR and prevent amyloid formationCitation11. This strategy seems to have a favourable effect on the progression of neuropathy, but has no effect on the progression of cardiac and ocular amyloidosis. Because of its limited applicability and the high risk of complications of transplantation, the development of alternative chemotherapeutic strategy is desirable. Less aggressive therapeutic strategies proposed for the prevention and treatment of amyloidosis include different types of approach: stabilization of tetrameric structure of TTR to prevent misfolding, modulation of the assembly process by stabilization of non-toxic amyloidogenic intermediates, and disruption of insoluble depositsCitation12–14.
Under physiological conditions, only small amount of T4 in the plasma is bound to TTR, so the vast majority of TTR T4 binding sites are unoccupied and available to bind small molecules that prevent tetramer dissociationCitation15. Recently tafamidis meglumine (Fx-1006A, trade name Vyndaqel) has received approval by the European Medicines Agency for the treatment of transthyretin amyloidosis in adult patients with stage-1 symptomatic polyneuropathy to delay peripheral neurologic impairment. Stabilization of the tetramer with tafamidis in the early stage seems to be a promising option for a therapeutic intervention. Currently a number of efforts are addressed to the synthesis of small molecules targeting the formation of toxic aggregates as a promising strategy to treat amyloidosis.
TTR is a homotetrameric β-sheet-rich protein composed of four monomers, namely A, B, C and D. Each subunit is composed of 127 amino acid and contains eight strands, A–H, seven to eight residues in lengthCitation16,Citation17. The two identical T4 binding site are located on the twofold symmetric axis at the interface between the two dimers. The two T4 molecules bind the tetramer with a negatively cooperative manner with two different binding constantsCitation18. The binding sites present a particular funnel-shaped morphology with polar residues located in the large outer cavities and in the bottom and a hydrophobic core placed at the center. TTR tetrameric structure is stable under physiological condition while moderately acid pH increases the rate of dissociation and aggregation of the proteinCitation19. Previous studies have shown that TTR misfolding leading to amyloid fibril formation can be prevented by compounds with very different scaffold which bind TTR binding siteCitation20. These molecules include several non-steroidal anti-inflammatory drugs (NSAIDs), among this class of compounds the most active TTR amyloid inhibitor is diflunisalCitation21,Citation22. In 2012, an international clinical study described the benefits of the use of diflunisal in TTR amylodosisCitation23.
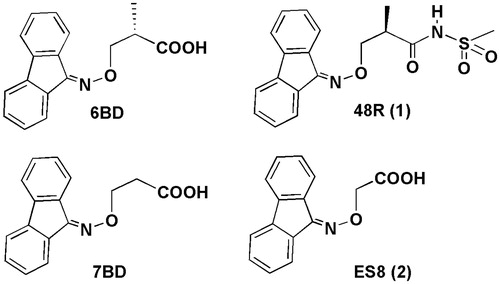
The X-ray analysis of the complex between the protein and the small molecules can be a valuable method to obtain useful information that can guide the design of new compounds able to stabilize the proteinCitation24. We report the results from the use of a new simplified procedure developed to obtain well-diffracting crystals of human transthyretin–ligand complexes without the need of a cumbersome soaking stepCitation25. The new approach avoids soak periods as long as 5 wk with a 10-fold molar excess of the ligand to achieve full saturation of both binding sitesCitation26. The method uses commercial protein and crystallization screens, yields crystals that are isomorphous with those for other complexes grown with ammonium sulphate or citrate based precipitants, within three days using less than six drops. Concerned that the use of non physiological high ionic strength crystallization and soaking conditions could bias the positioning of ligands, we analyzed the binding to TTR of (R)-3-(9-fluoren-9-ylideneaminooxy)-2-methyl-N-(methylsulfonyl) propionamide 48R(1) and 2-(9H-fluoren-9-ylideneaminooxy)acetic acid ES8(2) and at the same time we re-analyzed the binding of previously described fluorenyl derivatives 6BD, 7BDCitation27 () and report here our findings.
Figure 1. Chemical structures of fluorenone-based ligands.
Methods
Chemistry
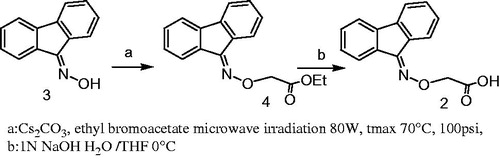
Compounds 6BD, 7BD and 1 were synthesized as reported in literatureCitation27. Compound 2 was synthesized starting from fluoren-9-one oxime (3) which was treated with ethyl bromoacetate to give the ester 4. The ester was hydrolyzed with a stoichiometric amount of 1 M NaOH solution to furnish the desired acid 2 (Scheme 1).
Scheme 1. Synthesis of compound 2.
Crystal growth and preparative steps
TTR ligand–crystal complexes have been obtained from various conditions from the Stura Footprint ScreenCitation28 from which crystals were obtained with high ionic strength precipitants and from crystallization conditions based on the high molecular weight polyethylene glycolCitation25. The latter conditions were used extensively in this study since large TTR-inhibitor complex crystals could be obtained in few hours with only three drops and made to grow to full size in three days or less. To induce nucleation and encourage crystal growth, a booster solution, consisting of NaCl was added at reservoirCitation25 and the drop immediately streak seeded. Since amylodogenesis inhibitors have low affinity for their target, 1 mM ligand was added as a precaution to the cryoprotectant so that during this brief soaking step that can last from 30 s to 2 min, the ligand does not wash out of the crystals. This strategy has allowed us to obtain crystallographic data at high resolution for TTR in complex with (S)-3-(9H-fluoren-9-ylideneaminooxy)-2-methylpropanoic acid ligand 6BD, with 3-(9H-fluoren-9-ylideneaminooxy)propanoic acid 7BD both reported previouslyCitation27 but also with (R)-3-(9H-fluoren-9-ylideneaminooxy)-2-methyl-N-(methylsulfonyl)propionamide (1)Citation27 with an amylodogenesis inhibition activity comparable to diflunisal and 2-(9H-Fluoren-9-ylideneaminooxy) acetic acid (2) ().
Table 1. Statistics for data collection, processing and refinement on TTR–ligand complexes.
Results
Crystallographic analysis
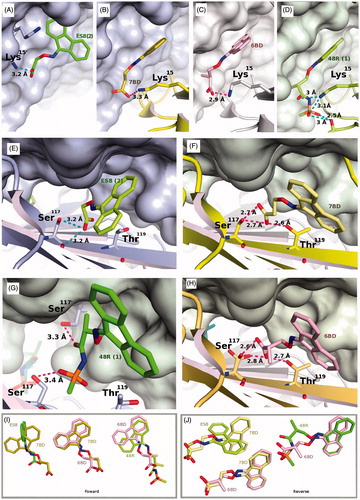
Crystals of the four inhibitors: 7BD, 6BD, 1 and 2 grown in polyethylene glycol belong to the orthorhombic space group P21212 with cell parameters comparable to those deposited in the PDB database from crystals grown at high ionic strength (). The resolution of the four structures ranges from 1.58 to 1.25 Å and there are no disordered portions of the protein although the region 99–104 can adopts two alternate conformations. The two TTR molecules in the asymmetric unit have only minor differences, and there are also small differences in the mode the ligands bind in the two TTR binding sites. In all cases the ligands show a dual binding mode: reverse and forward, so the carboxylic groups point alternatively towards the inner and the outer part of the binding pocket. Each alternative conformation is subjected to the symmetric crystallographic operation that builds the tetramer, so that the sum of the occupancies of the forward and binding modes of each inhibitor cannot exceed 0.5. The reverse mode is defined with the carboxylic moiety directed towards the inner binding pocket, interacting with Ser117 and Thr119 and the symmetric Ser117′ and Thr119′. In this mode, the fluorenyl ring is located at the outer binding pocket in hydrophobic cavity formed by Lys15 and the symmetric Lys15′, Ala108/108′, Leu17/17′, Thr106/106′ and Val121/121′. The fluorenyl ring maintains its overall position in the forward mode for 6BD, 7BD and 1, but the carboxylic group approaches Lys15. The dual binding mode remains valid for compound 2, but the positioning of the fluorenyl ring is at right angles to that of the other 3 compounds using an alternative hydrophobic pocket.
The electron density map can be interpreted with two molecules of inhibitor at each of the two TTR binding sites with similar disposition. 6BD binds both in reverse and forward binding mode orienting its carboxyl substituent towards the inner and outer binding pocket (). In the reverse mode, compound 6BD points the aliphatic linker toward the inner binding pocket with two conformations which simultaneously interact with HBP3/HBP3′ residues: Ser117/117′ and Thr119/119′. The two conformations have been assigned equal occupancy. The oxygens of carboxylic group are involved in hydrogen bonds with Ser117 (at distance of 2.6 Å and 2.8 Å; ) and with Thr119 (at distance of 2.7 Å). The fluorenyl ring is located into the hydrophobic pocket formed by Ala108/108′, Lys15/15′, Leu17/17′, Thr106/106′ and Val121/121′. In the forward mode the carboxylic group of aminooxy linker points toward the outer binding pocket making a hydrogen bond with Lys15 and 15′ (at distance 2.9 Å). The second inhibitor conformation is related by the crystallographic two-fold symmetry, so it makes identical interactions with the TTR binding site. The fluorenyl moiety is oriented into hydrophobic pocket.
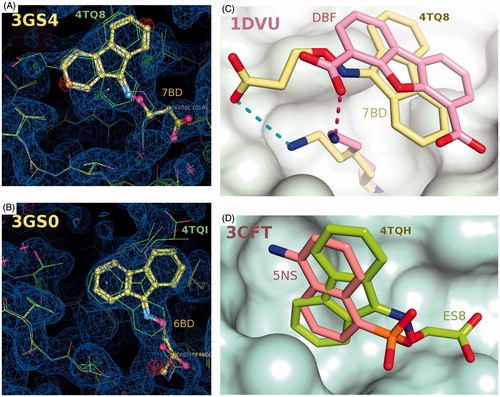
Figure 2. Schematic representation of the binding of ligands to TTR. All four ligands in this study bind at the crystallographic dimer interface of the TTR tetramer both in the forward and reverse mode. (A) Two orthogonal views of the electron density (weighted 2Fobs-Fcalc, contoured at 0.35, 1.35, 2.35, 3.35σ) for compound 6BD to show its positioning at each of the two binding sites in the TTR tetramer composed the a yellow/white dimer that forms the symmetric unit and its symmetric orange dimer. (B) Superposition of four 6BD molecules in their electron density. 6BD binding in the forward mode is shown in turquoise. The forward binding mode is characterized by the proximity of the carboxylic acid to Lys15. 6BD binding in the reverse mode is shown in light magenta. In this mode the carboxylic acid interacts with Ser117 and/or Thr119. Because 6BD is found at dimer interface in both the forward and reverse binding mode the electron density is the result of scattering contributions from four molecules of 6BD. The same is true for the other ligands. (C) The binding of 7BD to TTR resembles that of 6BD with only minor differences. The weaker electron density for the forward mode implies that either the carboxylate moiety is more flexible that in 6BD or that there is a preference for the reverse mode. (D) The fused ring system of the fluorenone of 6BD, 7BD and 48R (compound 1; ) occupy approximately the same position (top) while the ES8 (compound 2, ) fluorenone is positioned at right angles to the other three. (E) This profound change does not prevent ES8 from establishing interactions both in the forward and reverse modes, with the ring in the same position. (F) Electron density for 48R binding to TTR in both the forward and reverse binding modes. Stereo-images of ligand binding can be found in Supplementary Figure 2.
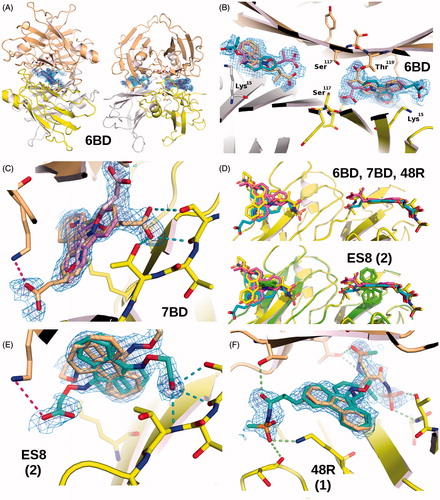
Figure 3. Details of the interactions established between ES8, 7BD, 6BD and 48R with TTR in the forward (A–D) and reverse binding modes (E–H). The various ligands are superimposed in pairwise manner in the insets, always in the same orientation in (A–D) or (E–H) to show the relative positioning of the ligands. (A) Binding of ES8. The shorter ligand adopts a drastically different binding strategy in positioning itself relative to Lys15 compared to 7BD, 6BD and 48R (panels B–D). This is achieved by using a totally different hydrophobic pocket compared to the other ligands. (B) Binding of 7BD in the forward mode. The fluorenone ring is positioned perpendicularly compared to ES8. (C) Binding of 6BD in the forward mode. The symmetric dimer is shown in surface representation to illustrate that there are substantial variations in the TTR internal surface due to the binding of different ligands that elicit compensatory side chain adaptations. (D) Binding of 48R to TTR in the forward mode, with the surface shown for the symmetric molecule. The methyl groups of 6BD and 48R occupy different pockets, this affects the surface of the symmetric dimer. These minor differences are likely to be important in tetramer stabilization. (E) Binding of ES8 in the reverse mode, interacting only with Ser117, while Thr119 must adopt a different rotamer to avoid a steric clash. (F) In the reverse binding mode the carboxylate of 7BD can establish interactions with both Ser117 and Thr119. (G) An interesting aspect of 48R is the ability to engage in interactions across the symmetry axis that relates the two dimers in the crystallographic unit cell, interactions that could contribute to tetramer stabilization. (H) Ser117 is shown as a superposition of three rotamers, a situation characteristic of this residue in several of the structures determined here. In the absence of a ligand, a water molecule often bridges an interaction between Ser117 and Thr119. All ligands need to displace this water molecule to be able to bind in the reverse mode. The interactions established by 6BD closely resemble those made by 7BD when both bind in the reverse mode. (I) Pairwise superposition of selected ligands to show the range of variability in ligand positioning within the binding site in the forward binding mode. (J) Pairwise comparison of ligand binding in the reverse mode.
The crystal of (R)-3-(9H-fluoren-9-ylideneaminooxy)-2-methyl-N-(methylsulfonyl) propionamide (1), (PDB ligand code = 48R) grew in the orthorhombic space group P21212 with cell parameters a = 43.1 Å, b = 86.1.Å, c = 63.5 Å. The cell parameters are almost identical to those of compounds 6BD and 7BD (). Compound 1 binds simultaneously at both hormone-binding sites either in the forward and reverse mode, either facing one or the other TTR monomer in the symmetry-related dimer, so that the electron density is the superposition of four alternative binding possibilities (). In reverse mode, the fluorenyl ring is located in outer binding and the sulfonamide linker points toward inner cavity. The oxygens of the sulphonamide group make hydrogen bond with Ser117/117′ (at distance 3.4 Å and 3.3 Å; ). In forward mode the sulphonamide linker interacts with Lys15/15′ with the amide oxygen at 2.9 Å and with the sulphonyl group at 3.5 Å. The two oxygens of sulphonamide moiety make hydrogen bonds with Ser52/52′.
The data for 2-(9H-Fluoren-9-ylideneaminooxy)acetic acid (2), (PDB ligand code = ES8) has been collected to 1.51 Å resolution and refined to Rvalue = 18.5% Rfree = 22.2% (). The parameters, a = 42.4 Å, b = 86.0 Å, c = 63.0 Å, differ from those for the other 3 ligands, mainly by having smaller a cell parameter. These differences may or may not be related to the different binding of the fluorenyl ring that for compound 2 binds at right angles compared to the other 3 ligands (). The inhibitor binds to both TTR-binding sites but with small variation in positioning. The electron density has been interpreted with a ligand is positioned in both TTR binding sites, either in the forward or reverse mode or in one of the two alternative conformations duplicated through the crystallographic symmetry operation (). This gives in total four alternative positions, corresponding to two conformations.
Crystals of 3-((9H-fluoren-9-ylideneamino)oxy)propanoic acid, 7BD, (PDB code: 4TQ8) grew in orthorhombic space group P21212 with cell parameters a = 43.1 Å, b = 85.9 Å, c = 63.4 Å, with a dimer in asymmetric unit (). Crystals diffracted to 1.52 Å resolution and data were refined to Rvalue = 18.6% Rfree = 21.0%. Two molecules of ligand bind TTR binding sites with similar orientations in monomers A and B. Two different alternative binding modes: reverse and forward, are adopted, so the carboxylic group may point both toward the inner or the outer binding pocket. In each binding mode, the inhibitor presents two symmetrical conformations with equal assigned occupancy (). In the monomer A both conformations are possible, so in reverse mode the carboxylic moiety heads towards the inner binding pocket and it interacts with Ser117 and Thr119 residues. The two carboxylic oxygen atoms form hydrogen bonds at distance 2.7 Å and 2.7 Å with Ser117 and at distance 2.6 Å to Thr119. At the two-fold axis, between the Ser117 and 117′ residues a water molecule can interact with the carboxylic moiety contributing to the hydrogen bond network within the inner binding pocket. This network connects the Ser117 residues of all four subunits helping maintain the tetrameric structure intact. The fluorenyl ring is located in the outer binding pocket in hydrophobic cavity composed by Lys15/15′, Ala108/108′, Leu17/17′, Thr106/106′ and Val121/121′. While in forward mode the carboxylic group points towards Lys15 (at distance 3.3 Å; 3 A) and the aromatic portion is disposed towards the inner binding pocket. Also, in monomer B the density map shows both reverse and forward binding modes. In reverse mode one oxygen of carboxylic group establishes hydrogen bond with Ser117 (at distance of 2.7 Å; ) and Thr119 (at 2.82 Å), while fluorenyl moiety is located in hydrophobic cavity.
TTR fibrillogenesis inhibition studies
The results of fibril formation assay for compounds 6BD, 7BD, 1 and 2 are compared to the reference drug diflunisal in ; the activity is reported as a percentage of inhibition of fibril formation. The 2-(9H-Fluoren-9-ylideneaminooxy)acetic acid (2) shows a percentage of inhibition of 49% and 77% at a concentration of 3.6 μM nd 7.2 μM, espectively. The inhibition values show that compound 2 maintains the capacity to inhibit the TTR fibril formation, even if with values slightly lower than compounds 6BD, 7BD and 1 are previously describedCitation27.
Table 2. In vitro acid-mediated wt-TTR (3.6 µM and 7.2 µM) fibrillogenesis inhibition of compounds 6DB, 7BD, 1 and 2.
Experimental section
Chemistry
Analytical reagents, starting materials and solvents were purchased from Sigma Aldrich (St. Louis, MO). Micro-wave-assisted reactions were run in a CEM Discover LabMate Microwave Synthesizer (Matthews, NC). 1H-NMR and 13C-NMR spectra were determined with Bruker 400 MHz or Varian Gemini 200 MHz spectrometers (Mountain View, CA). Chemical shifts (δ) are reported in parts per million (ppm) downfield from tetramethylsilane and referenced from solvent references. Coupling constants J are reported in Hertz; 13C-NMR spectra were fully decoupled. The following abbreviations are used: singlet (s), doublet (d), triplet (t), broad (br) and multiplet (m). Melting points were determined on a Köfler hotstage apparatus (Arthur H. Thomas Co., Philadelphia, PA) and are uncorrected. Chromatographic separations were performed on silica gel columns by flash column chromatography using Merck silica gel (70–230 mesh) (Darmstadt, Germany). Reactions were monitored by thin-layer chromatography (TLC) on Merck 0.2 mm pre-coated silica gel aluminium sheets (60 F-254) sheets were visualized under a UV lamp. Evaporation was performed in vacuo (rotating evaporator). Sodium sulphate was always used as the drying agent. Elemental analyses were performed by our Analytical Laboratory and within ± 0.4%.
Fluoren-9-one oxime (3)
The title compound was synthesized following the procedure described in literatureCitation33. The crude product was purified by crystallization from EtOH. (80% yield); m.p.: 184–186 °C (litt. 198 °CCitation34; 1H-NMR (400 MHz, DMSO-d6): 12.52 (s, 1H), 8.34–8.23 (d, 1H, J = 7.5 Hz), 7.88–7.82 (dd, 2H, J = 7.4 Hz, J = 17.24 Hz), 7.70–7.69 (d, 1H, J = 7.4 Hz), 7.51–7.32 (m, 4H). Calculated for C13H9NO: C, 79.98; H, 4.52; N, 7.17. Found: C, 79.85; H, 4.45; N, 7.09.
Ethyl 2-(9H-fluoren-9-ylideneaminooxy)acetate (4)
To a suspension of fluoren-9-one oxime (0.515 mmol) in CH3CN (2 mL) was added anhydrous Cs2CO3 (0.67 mmol) and ethyl bromoacetate (0.67 mmol). The suspension was submitted to microwave irradiation at a power of 80 W, tmax 70 °C, 100 psi and monitored by TLC. The reaction mixture was poured into ice, extracted with Et2O and washed with an aqueous 1 N NaOH solution. Organic layer dried and evaporated under vacuum gave the crude ester that was purified by flash chromatography (n-hexane:EtOAc 90:5). Evaporation of the fractions containing the main product yield pure 4. (78% yield); 1H-NMR (200 MHz, CDCl3): 8.39–8.35 (d, 1H, J = 7.5 Hz), 7.74–7.20 (m, 7H), 4.93 (s, 2H), 4.34–4.23 (q, 2H, J = 7.1 Hz), 1.34–1.27 (t, 3H, J = 7.1 Hz). Calculated for C17H15NO3: C, 72.62; H, 5.33; N, 4.98; Found: C, 72.68; H, 5.29; N, 4.88.
2-(9H-Fluoren-9-ylideneaminooxy)acetic acid (2)
1 N NaOH solution (1.11 mL) was added dropwise to a solution of ethyl 2-(9H-fluoren-9-ylideneaminooxy)acetate (3) (0.387 mmol) in H2O (2.22 mL) and THF (4.44 mL) at 0 °C. The mixture was stirred for 2 h at 0 °C and then at room temperature until the ester was completely hydrolyzed into acid. The reaction was monitored by TLC (n-hexane:AcOEt 8:2). After evaporation of the THF, the resultant solution was added with H2O and washed with Et2O. The water solution was acidified to pH 4 with 10% HCl, and extracted with CHCl3. Evaporation of the filtered organic layers afforded a crude yellow solid which was crystallized by n-hexane to give 2. (52%). m.p.: 225–226 °C, (litt. 237–239 °C dec.,Citation35; 1H-NMR (200 MHz, CDCl3): 8.30–8.26 (d, 1H, J = 7.5 Hz), 7.64–7.49 (m, 2H), 7.53–7.12 (m, 5H), 4.82 (s, 2H), 4.4 (br, 1H); 13C NMR (400 MHz, DMSO-d6,): 166.5, 148.2, 136.7, 135.7, 130.2, 127.7, 126.5, 125.5, 125.2, 124.6, 124.3, 117.4, 116.6, 116.6, 67.8. Found: C, 71.16; H, 4.33; N, 5.52. Calculated for C15H11NO3: C, 71.14; H, 4.38; N, 5.53.
Turbidimetric assay
Commercially available wild-type TTR (wt-TTR) was purchased from Calbiochem (Merck Millipore, Darmstadt, Germany). Compound 2 was dissolved in DMSO at a concentration of 7.2 mM provide a primary stock solution, the second stock solution was prepared in DMSO by 5-fold dilution of primary stock solution to a final concentration of 1.44 mM. 5 µM of this solution was added to 495 µL of a 7.2 µM WT-TTR solution in 10 mM phosphate (pH 7.6), 100 mM KCl, and 1 mM EDTA, buffer in a disposable plastic UV cuvette. The compound was incubated with TTR for 30 min at room temperature. After this period 500 µL of 200 mM acetate buffer (pH 4.2), 100 mM KCl, 1 mM EDTA was added to yield final TTR and inhibitor concentrations of 3.6:7.2 µM, respectively and a final pH of 4.4. The mixture was vortexed, then incubated at 37 °C for 72 h without stirring. After 3 days the solution was vortexed to redistribute any precipitate that had formed and the optical density was measured at 400 nm using a SPECTROstarNano (200–1000 nm) UV/Vis spectrophotometer (Ortenberg, Germany). The assays were performed in triplicate. Diflunisal was used as reference drug. The percentage of fibril formation was determined as previously describedCitation27. The turbidimetric assay, using equimolar concentrations of both inhibitor and TTR (3.6 µM), was carried out using a stock solution of inhibitor prepared in DMSO by 10-fold dilution of the primary stock solution (7.2 mM).
Crystallization experiments
The crystallization experiments were carried out by sitting drop vapour diffusion using 1 mg of lyophilized human TTR (Calbiochem, Merck Millipore, Darmstadt, Germany) dissolved in 100 μL 0.02% (w/v) NaN3 and dialysed against 0.1 M NaCl, 50 mM sodium acetate, pH 5.5, overnight. The drops were prepared with 1 μL protein solution and 1 μL reservoir solution using CrysChem plates equilibrated in a cooled incubator at 20 °C. Various alternative TTR crystallization conditions were obtained by screening using the Stura Footprint Screen (Molecular Dimensions Ltd., Cambridge, UK)Citation28. The most efficient crystallization strategy made use of various combinations of working solutions (WS) based on high molecular weight polyethylene glycols (PEG) mixed together to achieve small variation in pH and precipitating power following the principles of reverse screeningCitation36. The working solutions are taken directly from the commercial Stura Footprint Screen to avoid mixing errors. Typically, the reservoir solutions consisted of 70-60% of (WS1: 30% PEG-4000 (4 K), 0.2 M imidazole malate, pH 6.0) with 30% of (WS2: 12% MPEG-5 K, 0.1 M sodium acetate, pH 5.5) or 40% of (WS3: 20% PEG-4 K, 0.2 M imidazole malate, pH 6.0) or (WS4: 24% MPEG-5 K, 0.1 M sodium acetate, pH 5.5) or (WS5: 22.5% PEG-10 K, 0.1 M ammonium acetate, pH 4.5). Whenever crystals did not appear spontaneously the drops were streak seededCitation37. Since protein solubility varies from one ligand complex to another, 50 µL of a booster solution was added to the 500 µL the reservoir solution to achieve supersaturation if there was no response to the seeding. The two booster solutions used consisted of 5 M NaCl and 5 M NaCl with 0.2 M glacial acetic acid. After the boost, large crystals could be observed within a few hours. For data collection, the crystals were briefly soaked in cryoprotectant solution enriched with extra ligand before flash freezing.
The 3-((9H-fluoren-9-ylideneamino)oxy)propanoic acid (PDB ligand: 7BD)-TTR complex was prepared from TTR at 5 mg/mL and 10 mM fluorenyl derivative 7BD in DMSO, added in a ligand to protein volumetric ratio of 1:6. Crystals were obtained from 60% of working solution WS1 and 40% of either WS2 or WS5. The two solutions were thoroughly mixed together before the precipitant was added to the protein drop because of the high viscosity of the solutions. A single addition of the 5 M NaCl booster solution was sufficient for streak seeding to induce the growth of large crystals for X-ray diffraction.
(S)-3-((9H-fluoren-9-ylideneamino)oxy)-2-methylpropanoic acid (PDB ligand: 6BD) was dissolved in DMSO to make a 10 mM solution. The crystallization was carried out by TTR at 5 mg/mL and 6BD at 10 mM and 2 mM. The first experiment was made with protein ligand-solution containing 2 mM of inhibitor. The reservoir solution was prepared with 50% of WS1 and 50% WS3 thoroughly mixed. Large crystals were obtained after a single boost and streak seeding.
Other combinations of working solution were sampled to improve crystal quality. The new experiment was made with the 6BD concentration of 10 mM in DMSO. Crystals were obtained from 70% of WS1 and WS4. Large crystals were obtained after a single boost and streak seeding.
The TTR-ligand complex was prepared from TTR at 5 mg/mL and 10 mM (R)-3-(9-fluoren-9-ylideneaminooxy)-2-methyl-N-(methylsulfonyl) propionamide, compound 1, dissolved in DMSO. The reservoir condition used to obtained crystals was 60% of WS1 and 40% of WS2. Large crystals were obtained after a single boost and streak seeding.
The complex between TTR and 2-(9H-Fluoren-9-ylideneaminooxy)acetic acid, (2) (), was prepared from TTR at 5 mg/mL and 10 mM fluorenyl derivative in DMSO, added in a ligand to protein volumetric ratio of 1:6. Large crystals were obtained from 60–70% of working solution WS1 and 30–40% of either WS2 or WS4, respectively. The two solutions were thoroughly mixed together before the precipitant was added to the protein drop. A single addition of the 5 M NaCl booster solution was sufficient for streak seeding to induce the growth.
Data collection, structure determination and refinement
The procedure for cryoprotecting the crystals follows that described beforeCitation25,Citation38. Briefly, crystals were transferred to an XRL plate with micro-bridges with 6 µl mixed cryoprotectant and 0.5 µL ligand at 1 mM to avoid depletion of the ligand bound in the crystals during this short phase. After the cryosoak, the crystals were retrieved with a cryo-loop and plunged in liquid nitrogen in ESRF or Universal pucks. The crystals stored in a Dewar of liquid nitrogen were analyzed by X-ray diffraction at synchrotron facilities, namely at the ESRF (beam lines ID23-1 and ID23-2) in Grenoble and on beam line Proxima-2 at the Soleil storage ring in Saclay. Automatic data processing available at the ESRF synchrotron facilitiesCitation39 was used or the data was processed using XDSCitation40 with the “xdsme” script. The high-resolution limit for the data cut-off takes into account the data quality correlation coefficient CC1/2 (>35%) and the mean I/σ(I) (2 > x > 0.7) for the highest resolution shell to avoid discarding useful dataCitation31. The structures were solved by rigid body refinement using REFMAC5Citation41 starting with a TTR dimer in the P21212 with similar cell parameters without inhibitor. COOTCitation42 was used to interpret the electron density maps and to position the inhibitor built using the monomer library sketcher from CCP4 program suiteCitation43 in the difference electron density. The structure was subjected to at least three cycles of rebuilding and refinement with REFMAC5 and phenix.refineCitation32.
Comparison with previous PDB depositions
The option “Fetch PDB & Map using EDS”Citation44 in the program COOTCitation42 was used to compare the electron density maps of previously deposited structures with the model obtained with the same compounds crystallized with high molecular weight PEG. The models were aligned using the secondary structure matching algorithm (SSM)Citation45 implemented in COOT and the electron density viewed at 0.125 e/Å3.
Discussion
Inhibition of TTR amyloid formation has been linked to the binding of ligands in the inter-dimer interface, stabilizing the tetrameric assembly. The chemical characteristics of the inhibitors are highly variable, although some common motifs have emerged. The 9-fluorenyl derivatives described here follow the general trend by having an amphipathic character. While in most studies, inhibitor complex crystals with wild-type TTR have been prepared mainly using high ionic strength precipitants such as 1.4 M sodium citrateCitation46 or 2 M ammonium sulphateCitation27,Citation47, we have resorted to an alternative crystallization approach involving high molecular weight PEG. This approach has been used to grow crystals of certain TTR variants, such as the V30M, prevalent is FAP, with PEG 400Citation48. The results obtained here cannot fully validate the hypothesis that because high ionic strength conditions could shield the charge on Lys15, high salt crystallization skews the results in favour of the reverse binding mode. The preference for high molecular weight rather than the low molecular weight forms of polyethylene glycol derives from reverse screeningCitation36. The preference for the longer polymers is of secondary importance in this application compared to the contribution of PEG towards ligand solubilisation and TTR complex formation. The new approach to TTR co-crystallization avoids long soak periods needed to force ligands that are poorly soluble in high salt solutions through the crystalline lattice so as to reach the cavity at the interior of the TTR tetramer. The electron density in proximity of Lys15 within the TTR binding site for inhibitors 6BD, 7BD obtained in our study, shows in all cases a dual binding mode. Analysis of the experimental electron density maps (PDB entries: 3GS0, 3GS4; and ) obtained for the same two compounds under high ionic strength precipitants shows important differences only for 6BD (), suggesting that the crystallization does influence the results. Under high salt crystals both inhibitors 6BD and 7BD are oriented only in the reverse mode, while the electron density for the same molecules crystallized with the high molecular weight polyethylene glycol mixtures can be interpreted better by the superposition of four species: one in the forward mode, one in the reverse mode and their two symmetric pairs. In the reverse mode the carboxylic group interacts with Thr119, Thr119′, Ser117, Ser117′ and in the forward mode with Lys15, Lys15′. The fluorenyl moiety remains positioned in the hydrophobic binding cavity, in approximately the same manner in both the forward and reverse modes in good agreement with the docking analysisCitation27. One factor that could have influenced our ability of visualizing and interpreting both forward and reverse conformations could be the results of higher resolution diffraction data that in the previous study. However, the most important result stems from the structure of the new TTR complexes obtained in this study that show the separation between the hydrophobic and acidic portions of the inhibitor influences which hydrophobic binding site is exploited by the ligand. While the shorter ES8 analogue occupies an alternative hydrophobic binding pocket, the longer 48R shares the same hydrophobic binding pocked as 6BD and 7BD. All the ligands analysed here bind alternatively in the forward and reverse modes.
Figure 4. Comparisons of the binding of 7BD, 6BD and ES8 with other TTR crystal structures. (A) Superposition of the 4TQ8 and 3GS4 (24) on the experimental electron density for the latter. There is electron density in this structure that could support reverse and forward binding. (B) Superposition of the 4TQI and 3GS0 (27) (on the experimental electron density for the latter. It is clear that 6BD in 3GS0 binds exclusively in the reverse mode. (C) Superimposition of the ligand DBF from the human TTR in complex with dibenzofuran-4,6-dicarboxylic acid from PDB entry 1DVU. (26) The fused aromatic rings occupy similar positions in the two structures while the position of the carboxylate varies. (D) The binding pocket used by ES8, at right angles to that used by 6BD, 7BD and 48R.
Conclusions
The X-ray structures of TTR in complex with the acetic derivative ES8 compared with 7BD shows that a single atom in the linker between the tricyclic ring and acidic moieties can change the choice in positioning of the tricyclic ring. The same hydrophobic pocket is occupied by 6BD, 7BD and 48R but, the shorter linker allows the fused ring to flip and occupy an alternative pocket at right angles. This pocket has been described previously for 1-amino-5-naphthalene sulfonate (5NS) complex with TTRCitation49 where the distance between the hydrophobic-fused bicyclic ring and the acidic group is even shorter than that for ES8. The two orientations of fluorenyl ring seem to be influenced both by the length and the type of linker that joins the aromatic portion to the acidic moiety. The use PEG instead of ammonium sulphate or citrate may have contributed to the discovery that all 9-fluorenyl-like molecules orient themselves in the binding site, not only in reverse as previously reported, but also in the forward mode. This is emphasized by the presence of the ligand–Lys15 interaction.
The crystallographic studies have allowed us to better evaluate the potential of the 9-fluorenyl moiety as a privileged chemical scaffold for the development of new TTR fibril formation inhibitors with a mono-aryl structure. These information are valuable to guide the future design of new fluorenyl compounds in which appropriate decorations of the 9-fluorenyl scaffold, with groups of different nature, will allow the exploration of the steric requirements for effective ligand interaction in the hydrophobic binding cavity of TTR.
Supplementary material available online
Supplementary Figures 1 and 2
IENZ_1070265.pdf
Download PDF (8.4 MB)Acknowledgements
The authors thank Dr. Palaninathan Satheesh for his helpful in turbidimetric assays procedure.
Declaration of interest
The help of the staff at the ESRF and Soleil synchrotrons and the grant of beamtime are acknowledged with gratitude. This work was partially supported by Ministero dell’Istruzione, dell’Università e della Ricerca of Italy (PRIN 20109MXHMR_007).
Related Research Data
References
- Soprano DR, Herbert J, Soprano KJ, et al. Demonstration of transthyretin mRNA in the brain and other extrahepatic tissues in the rat. J Biol Chem 1985;260:11793–8
- Martone RL, Herbert J, Dwork A, Schon EA. Transthyretin is synthesized in the mammalian eye. Biochem Biophys Res Commun 1988;151:905–12
- Landers KA, Mortimer RH, Richard K. Transthyretin and the human placenta. Placenta 2013;34:513–17
- Chanoine JP, Alex S, Fang SL, et al. Role of transthyretin in the transport of thyroxine from the blood to the choroid plexus, the cerebrospinal fluid, and the brain. Endocrinology 1992;130:933–8
- Wojtczak A, Cody V, Luft JR, Pangborn W. Structures of human transthyretin complexed with thyroxine at 2.0 Å resolution and 3′,5′-dinitro-Nacetyl-L-thyronine at 2.2 Å resolution. Acta Crystallogr D Biol Crystallogr 1996;52:758–65
- Westermark P, Sletten K, Johansson B, Cornwell GG 3rd. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci USA 1990;87:2843–5
- Saraiva MJ. Transthyretin mutations in health and disease. Hum Mutat 1995;5:191–6
- Buxbaum JN, Tagoe CE. The genetics of the amyloidoses. Annu Rev Med 2000;51:543–69
- Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science 2003;299:713–16
- Sekijima Y, Wiseman RL, Matteson J, et al. The biological and chemical basis for tissue-selective amyloid disease. Cell 2005;121:73–85
- Pilato E, Dell’more A, Botta L, Arpesella G. Combined heart and liver transplantation for familial amyloidotic neuropathy. Eur J Cardio-Thoracic Surg 2007;32:180–2
- Connelly S, Choi S, Johnson SM, et al. Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses. Curr Opin Struct Biol 2010;20:54–62
- Saraiva MJ. Hereditary transthyretin amyloidosis: molecular basis and therapeutical strategies. Expert Rev Mol Med 2002;4:1–11
- Almeida MR, Gales L, Damas AM, et al. Small transthyretin (TTR) ligands as possible therapeutic agents in TTR amyloidoses. Curr Drug Targets CNS Neurol Disorders 2005;4:587–96
- Ong DS, Kelly JW. Chemical and/or biological therapeutic strategies to ameliorate protein misfolding diseases. Curr Opin Cell Biol 2011;23:231–8
- Blake CC GM, Oatley SJ, Rerat B, Rerat C. Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 Å. J Mol Biol 1978;121:339–56
- Wojtczak A. Crystal structure of rat transthyretin at 2.5 A resolution: first report on a unique tetrameric structure. Acta Biochim Pol 1997;44:505–17
- Andrea TA, Cavalieri RR, Goldfine ID, Jorgensen EC. Binding of thyroid hormones and analogues to the human plasma protein prealbumin. Biochemistry 1980;19:55–63
- Foss TR, Wiseman RL, Kelly JW. The pathway by which the tetrameric protein transthyretin dissociates. Biochemistry 2005;44:15525–33
- Nencetti S, Orlandini E. TTR fibril formation inhibitors: is there a SAR? Curr Med Chem 2012;19:2356–79
- Miller SR, Sekijima Y, Kelly JW. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Investig 2004;84:545–52
- Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 2006;13:236–49
- Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013;310:2658–67
- Palaninathan KS. Nearly 200 X-ray crystal structures of transthyretin: what do they tell us about this protein and the design of drugs for TTR amyloidoses? Curr Med Chem 2012;19:2324–42
- Ciccone L, Tepshi L, Nencetti S, Stura EA. Transthyretin complexes with curcumin and bromo-estradiol: evaluation of solubilizing multicomponent mixtures. N Biotechnol 2015;32:54–64
- Klabunde T, Petrassi HM, Oza VB, et al. Rational design of potent human transthyretin amyloid disease inhibitors. Nat Struct Biol 2000;7:312–21
- Palaninathan SK, Mohamedmohaideen NN, Orlandini E, et al. Novel transthyretin amyloid fibril formation inhibitors: synthesis, biological evaluation, and X-ray structural analysis. PLoS One 2009;4:e6290
- Stura EA, Nemerow GR, Wilson IA. Strategies in the crystallization of glycoproteins and protein complexes. J Crystal Growth 1992;122:273–85
- Vera L, Stura EA. Strategies for protein cryocrystallography. Crystal Growth Design 2014;14:427–35
- Newman J. Novel buffer systems for macromolecular crystallization. Acta Crystallogr D Biol Crystallogr 2004;60:610–12
- Diederichs K, Karplus PA. Better models by discarding data? Acta Crystallogr D Biol Crystallogr 2013;69:1215–22
- Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D. 2010;66:213–21
- Pan HL, Fletcher TL. Derivatives of fluorenone XXX. Rearrangement and antitumor activities of some 9-oxofluorene oximes. 6(5H)-phenanthridones. J Med Chem 1969;12:822–5
- Anet FAL, Bavin PMG, Dewar MJS. The reaction of nitromethane with fluorenone and benzophenone in polyphosphoric acid. Can J Chem 1957;35:180–2
- Richardson A Jr. Aminooxyacetic acid derivatives. J Med Chem 1964;7:824–6
- Stura EA. Strategy 3: reverse screening. In: Bergfors TE, ed. Crystallization of proteins: techniques, strategies and tips. A laboratory manual. La Jolla (CA): International University Line. 1999:113–24
- Stura EA, Wilson IA. Applications of the streak seeding technique in protein crystallization. J Cryst Growth 1991;110:270–82
- Ciccone L, Vera L, Tepshi L, et al. Multicomponent mixtures for cryoprotection and ligand solubilization. Biotechnol Rep 2015;7:120–7
- Beteva A, Cipriani F, Cusack S, et al. High-throughput sample handling and data collection at synchrotrons: embedding the ESRF into the high-throughput gene-to-structure pipeline. Acta Crystallogr D Biol Crystallogr 2006;62:1162–9
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr 2010;66:133–44
- Murshudov GN, Skubak P, Lebedev AA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 2011;67:355–67
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010;66:486–501
- Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 2011;67:235–42
- Kleywegt GJ, Harris MR, Zou JY, et al. The Uppsala electron-density server. Acta Crystallogr D Biol Crystallogr 2004;60:2240–9
- Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 2004;60:2256–68
- Moses AC, Rosen HN, Moller DE, et al. A point mutation in transthyretin increases affinity for thyroxine and produces euthyroid hyperthyroxinemia. J Clin Investig 1990;86:2025–33
- Green NS, Palaninathan SK, Sacchettini JC, Kelly JW. Synthesis and characterization of potent bivalent amyloidosis inhibitors that bind prior to transthyretin tetramerization. J Am Chem Soc 2003;125:13404–14
- Trivella DB, Bleicher L, Palmieri LC, et al. Conformational differences between the wild type and V30M mutant transthyretin modulate its binding to genistein: implications to tetramer stability and ligand-binding. J Struct Biol 2010;170:522–31
- Lima LMTR, Silva VDA, Palmieri LDC, et al. Identification of a novel ligand binding motif in the transthyretin channel. Bioorg Med Chem 2010;18:100–10