
Abstract
Background: The prevalence of capillary malformations, also known as port-wine stains (PWS), is 0.3%. Familial segregation can occur. The capillary malformation–arteriovenous malformation (CM-AVM) phenotype is caused by mutations in the RASA1 gene. In PWS familial cases, the inheritance is considered to be autosomal dominant with variable penetrance. Objective: Investigation of the heredity of PWS among patients who attended the vascular anomaly section at the Department of Dermatology in Malmoe, Southern Sweden, between 1993 and 2004 and to study the involvement of the RASA1 gene in patients with a positive family history of PWS. Subjects and methods: A total of 254 patients were examined and given a questionnaire regarding family history of PWS. The first group of 175 patients (109 females and 66 males) reported a negative family history. The other group of 65 patients (46 females and 19 males) reported a positive family history (50% parents or brothers and sisters). Results: The heredity of PWS was 27% (65/240). Twenty-one patients with a positive family history and relatives had no CM-AVM phenotype for mutations in the RASA1 gene. Conclusion: PWS may have a stronger heredity component than it was reported earlier and inheritance should be considered when counseling a patient. RASA1 mutations do not explain the PWS in our patients.
Introduction
Port-wine stains (PWS) are capillary malformations, the most common congenital, slow-flow vascular malformations of the skin. PWS are composed of dilated dermal capillaries, the so-called “capillary-venular-like channels” (Citation1,Citation2). There are no differences in vessel number, but there is an altered or even absent neural modulation of the vascular plexus with a decrease in both sympathetic and sensory innervations of the papillary plexus (Citation3,Citation4). The prevalence of PWS is stated to be 0.3–0.6% in newborns. The male-to-female ratio is 1:1 (Citation1,Citation5). Although generally not considered a familial condition, earlier studies have reported an inheritance of 8–22% (Citation6–9). The segregation pattern is often compatible with autosomal dominant inheritance, as for capillary malformation–arteriovenous malformation (CM-AVM). For the first time, CM-AVM was described to be caused by heterozygous mutations in the RASA1 gene on chromosome 5q13.1–14.3 (Citation10–12). These mutations cause abnormal angiogenic remodeling of the primary capillary plexus (Citation13). In CM-AVM, patients present with multifocal cutaneous capillary malformations often in association with fast-flow lesions, such as cutaneous, subcutaneous, intramuscular, intraosseous, cerebral, and spinal arteriovenous malformations or arteriovenous fistulas. Some patients with CM-AVM have Parkes Weber syndrome, a large congenital cutaneous vascular stain in an extremity, with bony and soft tissue hypertrophy and microscopic arteriovenous shunting. Previously, arteriovenous malformations and arteriovenous fistulas have been considered non- hereditary (Citation13). Our aim was to investigate the heredity of PWS among patients who attended the laser and vascular anomaly section at the Department of Dermatology in Malmoe, Southern Sweden, between 1993 and 2004 and the involvement of the RASA1 gene in patients with a positive family history of PWS.
Materials and methods
Study population
The retrospective study was a collaboration between the Department of Dermatology, UMAS laser clinic in Malmoe, Sweden and the Laboratory of Human Molecular Genetics, de Duve Institute, Université Catholique de Louvain, Brussels, Belgium. Patients attending the laser and vascular anomaly section at the Department of Dermatology in Malmoe between 1993 and 2004 were examined by a trained specialist for PWS. The study was approved by the local ethics committee at Lund University 490-02. Details of clinical examination were recorded with special reference to site, color, and texture of the PWS, and pictures were taken after informed consent was obtained. After clinical examination, patients were given a questionnaire, in which they were asked whether they knew any relative who had a red birthmark like themselves and, if so, where it was located. After returning the questionnaires, patients and relatives with a positive family history were selected, interviewed, and blood samples were obtained from an antecubital vein of the patients for screening for RASA1 gene mutations.
RASA1 screening
To amplify all 25 exons, including exon–intron boundaries, 26 sets of primers were designed. Genomic DNA was screened by denaturing high-performance liquid chromatography on the WAVE 3500 HS system (Transgenomic Inc., Omaha, NE, USA). Fragments with abnormal profiles were sequenced on a Beckman capillary sequencer. Numbering of nucleotides was based on the cDNA, sequence NM_002890.1, with the adenosine of the ATG start codon marked as + 1.
Results
Positive and negative family history groups
A total of 254 patients were examined and given a questionnaire inquiring about a family history of PWS (). During the initial review of the returned questionnaires and assessment of patients, 14 patients were excluded due to diagnosis other than PWS. In the negative family history group, a total of 11 patients were excluded with the following diagnosis: 4 hemangiomas, 3 Klippel–Trenaunay syndromes (KTS), 3 nevi, and 1 lichen sclerosus et atrophicus.
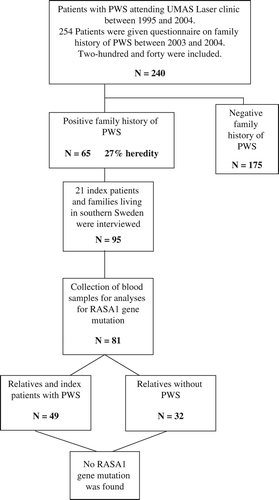
In the group with positive family history, a total of four patients were excluded because two had KTS, one had a hemangioma, and one had blue rubber bleb nevus syndrome. Hence, 240 patients were included and divided into two groups. The first group consisted of 65 subjects who had a positive family history of PWS (): 19 males (median: 25 [range: 2–53] years) and 46 females (median: 24 [range: 1–63] years). Clinical examination of the patients was done by the investigators. The male-to-female ratio and distribution of PWS was 1:2.4. In the other group, 175 subjects had a negative family history of PWS (): 66 males (median: 30 [range: 1–77] years) and 109 females (median: 27 [range: 2–74] years). The male-to- female ratio and distribution of PWS was 1:1.7. In the positive family history group, the most common location of the PWS was in the following order: face (74%), neck (22%), trunk (15%), legs (11%), arms (8%), and hands (2%). The group with the negative family history had similar location: face (79%), neck (20%), trunk (16%), legs (9%), arms (7%), and hands (6%).
Table I. All characteristic for the group.
Table II. All characteristic for the group.
The lesions were clinically presented mainly as flat, confluent, patchy, and pink/red in both groups. On the basis of these groups, the heredity of PWS can be estimated to be 27% (65:240).
We were interested in seeing whether the hypertrophic PWS could be different in comparison with macular PWS regarding the genetics. We found that 7% in the positive family history group had hypertrophic skin surface, . In the negative family history group, 20% had hypertrophic skin surface.
The RASA1 gene-screening group
From the group with positive family history, 21 subjects (8 males: 13 females) and their family members were included in the RASA1 gene screens (). A total of 95 subjects were included. Out of 95 subjects, 59 male (n = 27):female (n = 32) had a PWS (62% of the entire group). We were able to obtain 81 blood samples for analysis. Some family members were deceased or did not agree to give a blood sample (). In this group, 49 subjects including the index patients had a PWS: 20 males (median: 34 [range: 10–79] years) and 29 females (median: 35 [range: 7–89] years). The male-to-female ratio and distribution of PWS was 1:1.4. The characteristics of all 49 with a PWS are given in . The lesions were most commonly located in the following areas: face (41%), neck (41%), trunk (10%), arms (14%), legs (12%), and hands (4%). A majority of lesions clinically presented as flat, confluent, and pink patches. We did not identify any RASA1 gene mutation in our patients after the screening.
Table III. All characteristic for the group.
Discussion
In this retrospective questionnaire-based study, we showed a familial tendency of common CM for 65 out of 240 patients, which translates to a 27% heredity for PWS. In the group with positive family history, 21 patients and 60 relatives were screened for RASA1, mutations which cause the familial CM-AVM phenotype. We did not identify any mutation in our patients. Earlier studies have reported a prevalence of 8–22% (Citation6–9). These data suggest that PWS may have a higher heredity component in our selected population. In the literature, the male-to-female ratio is reported to be equal (Citation1–2). We found a higher female preponderance in the negative (62%) and positive (70%) family history groups. However, this was also the case for the RASA1-screened group (59%), in spite of the elimination of selection bias due to random selection of our 21 index subjects and family members. The above findings and the frequencies regarding site, color, pattern, and skin surface of the PWS studied in all three groups are similar to those of previous studies, with a trend for developing darker and more hypertrophic PWS with age (Citation6,Citation8,Citation9). The percentage of patients with PWS in whom skin thickening and nodularity will develop has been earlier estimated to be 10% (Citation6). In our study, we found a frequency of 4–14% of hypertrophic papular lesions in all three groups primarily located on the face. The onset of this thickening has been reported to occur in the 20–39-year-old group, with continued progression throughout the life of the affected individual (Citation14).
Sanchez-Carpintero et al. (Citation15) suggested that the reason why the face compared with truncal and extremity lesions is the most common site of PWS and associated thickening may be related to the fact that cranial mesenchyme, unlike the rest of the body, is derived from the neural crest rather than the mesoderm. Additionally, the well-recognized distribution of facial PWS in a segmental pattern corresponds to one or more branches of the trigeminal nerve.
Sanchez-Carpintero et al. (Citation15) also reported a late-stage PWS on the face of a 75-year-old man with wide epithelial and mesenchymal changes that were not restricted to focal tumor-like nodules. They suggested a multigermline developmental field defect in the pathogenesis of this lesion, perhaps mediated by somatic mutation. It is becoming more evident that genetic factors play a critical role in the pathogenesis of vascular anomalies. They are classified into vascular tumors and vascular malformations (Citation16). Vascular malformations are usually present at birth, grow proportionally with the patient, and rarely regress. These malformations include capillary, venous, arterial, and lymphatic telangiectasia, and combined or mixed vascular malformations.
In contrast to vascular tumors (e.g., hemangiomas), molecular genetic studies have identified disease-causing genes for a variety of vascular malformations. Hereditary venous malformation capillary malformation (VMCM) are caused by dominant TIE2 mutations (Citation17–18) and glomuvenous malformations are caused by loss-of-function mutations in glomulin (Citation19).
For the PWS, a genetic locus, CMC1, has been mapped to chromosome 5q14–21 in a study of 13 families (Citation10). Subsequent studies identified the RASA1 gene, which encodes a p120-RasGTPase-activating protein (p120-RasGAP). The function of p120-RasGAP is to switch the active GTP-bound Ras to the inactive GDP-bound form, and RASA1 defects probably cause abnormal angiogenic remodeling of the primary plexus (Citation15). RASA1 mutations have been detected in families with CM-AVM associated with arteriovenous malformation, arteriovenous fistula, or Parkes Weber syndrome (Citation11,Citation20). Four of the six families investigated were identified in Belgium and 39 individuals carried a RASA1 mutation. The CM of CM-AVMs were clinically small, round-to-oval, pink-red, typically multiple, and in a haphazard distribution (Citation13). Similar clinical findings have been seen in other larger follow-up series (Citation21). In our study only 3 subjects had more than 3 lesions in the affected group. Usually, PWS are sporadic and there is an incomplete penetrance (Citation10).
Conclusions
Our data show that there is a stronger heredity component of common PWS than earlier reported (in our selected population); however, we did not detect a RASA1 gene mutation in these lesions (PWS). Further investigations are warranted to locate genetic markers for PWS.
Contributors
Agneta Troilius was the main investigator who performed the study and wrote most of the paper. Edgar Lauritzen collected the blood samples and obtained funding and approval from the local ethics committee. Miikka Vikkula obtained funding and Nicole Revencu performed the genetic analyses. Bo Ljunggren and Åke Svensson participated in planning of the study, obtained funding, and supervised the study. All authors contributed to data evaluation and preparation of the final paper.
Acknowledgements
The authors are grateful to all family members for their participation in the study. The work was supported by the Edvard Welander Foundation (to EL), and the Interuniversity Attraction Poles initiated by the Belgian Federal Science Policy, P7/43, as well as the F.R.S.-FNRS (Fonds de la Recherche Scientifique) (both to MV). The authors thank June Palmkvist for secretarial help.
Declaration of interest: The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
References
- Jacobs AH, Walton RG. The incidence of birthmarks in the neonate. Pediatrics. 1976;58:218–222.
- Barsky SH, Rosen S, Geer DE, Noe JM. The nature and evolution of port wine stains: a computer-assisted study. J Invest Dermatol. 1980;74:154–157.
- Smoller BR, Rosen S. Port-wine stains. A disease of altered neural modulation of blood vessels? Arch Dermatol. 1986;122:177–179.
- Rydh M, Malm M, Jernbeck J, Dalsgaard CJ. Ectatic blood vessels in port-wine stains lack innervation: Possible role in pathogenesis. Plast Reconstr Surg. 1991;87:419–422.
- Osburn K, Schosser RH, Everett MA. Congenital pigmented and vascular lesions in newborn infants. J Am Acad Dermatol. 1987;16:788–792.
- Mills CM, Lanigan SW, Hughes J, Anstey AV. Demographic study of port wine stain patients attending a laser clinic: family history, prevalence of naevus anaemicus and results of prior treatment. Clin Exp Dermatol. 1997;22:166–168.
- Van der Horst CMAM, van Eijk TGJ, de Borgie CAJM, Koster PHL, Struycken PM, Strackee SD. Hereditary port-wine stains, do they exist? Lasers Med Sci. 1999;14:238–243.
- Troilius A. Characterisation and treatment of patients with port wine stains with special reference to the emotional impact. PhD thesis, Lund University, Lund, Sweden, 1999.
- Troilius A, Wrangsjo B, Ljunggren B. Patients with port-wine stains and their psychosocial reactions after photothermolytic treatment. Dermatol Surg. 2000;26:190–196.
- Eerola I, Boon LM, Watanabe S, Grynberg H, Mulliken JB, Vikkula M. Locus for susceptibility for familial capillary malformation (‘port-wine stain’) maps to 5q. Eur J Hum Genet. 2002;10:375–380.
- Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, Watanabe S, et al. Capillary malformation–arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240–1249.
- Breugem CC, Alders M, Salieb-Beugelaar GB, Mannens MM, Van der Horst CM, Hennekam RC. A locus for hereditary capillary malformations mapped on chromosome 5q. Hum Genet. 2002;110:343–347.
- Boon LM, Mulliken JB, Vikkula M. RASA1: variable phenotype with capillary and arteriovenous malformations. Curr Opin Genet Dev. 2005;15:265–269.
- Klapman MH, Yao JF. Thickening and nodules in port-wine stains. J Am Acad Dermatol. 2001;44:300–302.
- Sanchez-Carpintero I, Mihm MC, Mizeracki A, Waner M, North PE. Epithelial and mesenchymal hamartomatous changes in a mature port-wine stain: Morphologic evidence for a multiple germ layer field defect. J Am Acad Dermatol. 2004;50:608–612.
- Enjolras O, Mulliken JB. Vascular tumors and vascular malformations (new issues). Adv Dermatol. 1997;13:375–423.
- Vikkula M, Boon LM, Carraway KL 3rd, Calvert JT, Diamonti AJ, Goumnerov B, et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181–1190.
- Limaye N, Wouters V, Uebelhoer M, Tuominen M, Wirkkala R, Mulliken JB, et al. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat Genet. 2009;41:118–124.
- Brouillard P, Boon LM, Mulliken JB, Enjolras O, Ghassibé M, Warman ML, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am J Hum Genet. 2002;70:866–874.
- Thiex R, Mulliken JB, Revencu N, Boon LM, Burrows PE, Cordisco M, et al. A novel association between RASA1 mutations and spinal arteriovenous anomalies. AJNR Am J Neuroradiol. 2010;31:775–779.
- Revencu N, Boon LM, Mulliken JB, Enjolras O, Cordisco MR, Burrows PE, et al. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat. 2008;29:959–965.