Abstract
AZD9668 is a fully reversible, selective, oral inhibitor of neutrophil elastase, a protease implicated in chronic obstructive pulmonary disease (COPD). Efficacy, safety and tolerability of AZD9668 (5, 20 and 60 mg bid) were compared with placebo in a randomised, double-blind, placebo-controlled, 12-week, Phase IIb trial (NCT00949975: approved by an Investigational Review Board), in patients with symptomatic COPD receiving maintenance tiotropium. The primary endpoint was pre-bronchodilator forced expiratory volume in 1 second (FEV1). Secondary endpoints included forced vital capacity and inspiratory capacity, peak expiratory flow, Breathlessness, Cough and Sputum Scale score, exercise capacity, quality of life (QoL), exacerbation assessments, safety and pharmacokinetics. Exploratory endpoints included inflammatory and tissue degradation biomarkers. A total of 838 patients were randomised to AZD9668 5 mg bid (212 patients), 20 mg bid (206 patients), 60 mg bid (202 patients) or placebo (218 patients). AZD9668 showed no effect on lung function, respiratory signs and symptoms, QoL or biomarkers. At end of treatment, the change in mean pre-bronchodilator FEV1 for AZD9668 60 mg bid compared with placebo was 0.00L (95% confidence interval: −0.05, 0.04; p = 0.873). Overall, AZD9668 was well tolerated; the numbers of patients with adverse events (AEs), serious AEs and AEs leading to discontinuation were similar in each of the four study groups.
AZD9668 60 mg bid showed no clinical benefit and no effect on biomarkers of inflammation or tissue degradation when added to tiotropium in patients with COPD. These results raise important questions for future investigation of anti-inflammatory and disease-modifying agents in patients with COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is a leading cause of morbidity and mortality worldwide. It is characterised by airflow limitation that is not fully reversible, associated with an abnormal inflammatory response of the lung to inhaled cigarette smoke or other noxious particles or gases (Citation1). Although existing medications decrease symptoms and/or complications, none have been shown to modify the long-term decline in lung function that is the hallmark of COPD. Neutrophils play a significant role in the chronic inflammation underlying respiratory diseases such as bronchiectasis, cystic fibrosis and COPD (Citation2).
The neutrophil numbers in the airways increases with Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage and is inversely related to the forced expiratory volume in 1 second (FEV1) (Citation3). Neutrophil elastase (NE), an enzyme stored predominantly within the azurophilic granules of neutrophils, has the capacity to degrade a broad spectrum of structural and immune regulatory proteins including elastin, fibronectin, collagen, α1-antitrypsin and tissue inhibitors of matrix metalloproteinases (Citation4). Compelling evidence for an imbalance between proteases, such as NE, and antiproteases in the lungs of patients with COPD (Citation1), has intensified the search for pharmacological agents, which could restore this balance. So far, the concept of NE inhibition has been clinically employed only in a limited context, by the use of sivelestat (approved in Japan), developed for the treatment of acute lung injury and acute respiratory distress syndrome (Citation5,6).
AZD9668 is a novel, orally bioavailable NE inhibitor. Preclinical studies have demonstrated that AZD9668 is a potent, fully reversible inhibitor, with at least 100-fold selectivity for human NE compared with other serine proteases and 100 other enzymes, receptors and ion channels (Citation7). Efficacy of oral administration of AZD9668 has been demonstrated in in vivo models of acute lung injury and in an acute smoke model in rodents (Citation7). Three double-blind, randomised, placebo-controlled Phase I/IIa studies have investigated the pharmacokinetics (PK), safety and tolerability of AZD9668 in 107 healthy Caucasian and Japanese volunteers and 18 patients with COPD using single and multiple doses of AZD9668 for up to 14 days (Citation8). AZD9668 was well tolerated at single doses up to 150 mg and multiple doses up to 70 mg. The short elimination half-life was consistent with bid dosing and steady state was reached by Day 2.
This was a proof-of-concept and a dose-finding study to investigate the short-term efficacy, safety and tolerability of AZD9668 in patients with COPD receiving background maintenance treatment with tiotropium. A parallel Phase IIb study investigated the efficacy, safety and tolerability of AZD9668 in patients with COPD treated with maintenance budesonide/formoterol.
Methods
Design
We conducted a 12-week, randomised, double-blind, placebo-controlled, parallel group, Phase IIb study (NCT00949975) to evaluate the efficacy and safety of AZD9668 administered orally at three dose levels to patients with COPD receiving treatment with tiotropium (). The study was conducted at 138 centres in 12 countries (Australia, Canada, Germany, Japan, Philippines, Poland, Republic of Korea, Russian Federation, Slovakia, Taiwan, Ukraine and the USA).
Figure 1. Study design.
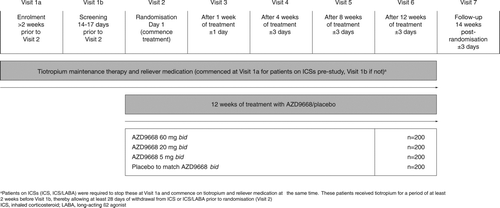
Eligible patients were randomised (1:1:1:1) using a computer-generated randomisation sequence to one of the four arms. Each patient was assigned a unique randomisation code strictly sequentially as patients became eligible for randomisation. The codes were computer-generated in blocks of 4. AZD9668 was provided as tablets for oral administration at strengths of 2.5, 10 or 30 mg. To ensure blinding, matching placebo tablets were identical in appearance. Patients in all treatment arms were to take the same dosage of investigational product i.e. 2 tablets bid (with doses approximately 12 hours apart) for 12 weeks. Treatment codes were not broken for the planned analyses of data until all decisions on the evaluation quality of the data from each individual patient were made and documented.
Patients
Patients were eligible for inclusion in the study if they were 40–80 years of age and had a smoking history of 10 pack-years or more, a diagnosis of COPD with symptoms for at least 1 year, a post-bronchodilator FEV1 of 40–80% of the predicted normal value, a ratio of FEV1 to forced vital capacity (FVC) of <70%, a history of at least one exacerbation treated with parenteral steroids and/or antibiotics within 4 weeks to 12 months before screening, and a total Breathlessness, Cough and Sputum Scale (BCSS) symptom score ≥2 per day for at least 7 of the last 14 days before Day 1. Further inclusion and exclusion criteria are detailed in the accompanying Supplementary Material.
Treatments
After a 2-week run-in period, eligible patients were randomised to receive AZD9668 (5, 20 or 60 mg bid) or placebo for 12 weeks. Intake of investigational product was recorded in a patient eDiary and was used as a measure of compliance. All patients received tiotropium maintenance medication (18 μg each morning via HandiHaler® inhaler [Boehringer Ingelheim]) during the run-in and treatment periods. Patients were provided with salbutamol sulphate 100 μg/dose as reliever medication or could continue to use their existing short-acting β2-agonist (SABA) reliever medication. All patients were instructed in the use of the inhaler devices. If patients were taking inhaled corticosteroids (ICS; including combinations with long-acting β2-agonists [LABA]), these were discontinued and tiotropium initiated at least 28 days before Day 1.
Assessments
The primary endpoint was the pre-bronchodilator FEV1 at clinic visits. Secondary endpoints were: post-bronchodilator FEV1, and pre- and post-bronchodilator FVC and inspiratory capacity (IC) measured at clinic visits; daily morning and evening peak expiratory flow (PEF) and FEV1 measured by patients at home; daily recordings of BCSS and EXAcerbations of Chronic pulmonary disease Tool (EXACT) scores, and use of reliever medication by patients at home; sputum colour (Bronkotest® 5-point colour scale) measured at clinic visits; exercise capacity (6-minute walk test) and health-related quality of life (QoL) (St George's Respiratory Questionnaire [SGRQ] and Functional Performance Inventory-Short Form [FPI-SF] - exploratory variable, completed in English speaking countries only) assessed at the clinic visits at Day 1 and Week 12; and clinic-defined exacerbations (a worsening in COPD requiring a course of antibiotics and/or systemic steroids and/or emergency room treatment and/or hospitalisation).
Safety endpoints included adverse events (AEs) (coded according to the medical dictionary for regulatory activities); laboratory evaluations, vital signs, 12-lead electrocardiogram and physical examinations. For PK assessment, blood samples were taken pre-dose and 0.5–4 hours post-dose at clinic visits at Weeks 4 and 12.
Exploratory endpoints included inflammatory biomarkers in pre-dose serum (normal C-Reactive Protein [n-CRP]) and plasma (interleukin [IL]-8, IL-6 and tumour necrosis factor-alpha [TNF-α]), and urinary biomarkers (creatinine-normalised desmosine), assessed at clinic visits at Day 1 and Weeks 4 and 12.
Statistical analyses
With a sample size of approximately 800 patients (200 patients in each treatment group), the study had 80% power to detect an effect on clinic FEV1 if the true difference was 62 mL, assuming a standard deviation of 250 mL, at the 5% significance level with a 1-sided test. The efficacy, safety and PK analysis populations comprised all patients who received at least one dose of study medication and for whom post-dose data for that population were available.
For data obtained at the clinic, the primary analysis compared end of treatment value (i.e. last value recorded for each patient, which is equivalent to the principle of last observation carried forward [LOCF]) between groups in an analysis of covariance (ANCOVA), with the baseline value (Day 1) as covariate and country and treatment as fixed factors.
For the primary endpoint only (end of treatment pre-bronchodilator FEV1 at clinic visits), a hierarchical decreasing procedure was planned; i.e., the analysis started with the highest dose level and proceeded to the lowest dose level, stopping when statistical significance was not met. As the study was exploratory in nature, a 2-sided p-value of <0.1 was considered significant. Data were summarised by visit using LOCF and plots and summaries showing the least squares means (LSM) difference and 95% confidence interval [CI] between AZD9668 and placebo were produced.
For diary card data, a similar ANCOVA analysis as for the clinic data was performed, but with baseline defined as the average of the last 10 days before randomisation and the value at the end of treatment as the average of the last available 6 weeks. Data were summarised by 4-weekly periods, with last observed period mean carried forward where necessary.
The time to first event-based COPD exacerbation was analysed using a Cox proportional-hazards model, stratified by country. Hazard ratios and 95% CI for the difference between AZD9668 and placebo were calculated and a Kaplan-Meier plot of time to first exacerbation during the randomised on-treatment period was constructed. All safety data were summarised descriptively for the safety analysis population. Biomarker data were analysed on a log scale using ANCOVA. The results were back transformed to give the ratio of the biomarker in patients on AZD9668 to those on placebo (no adjustment for multiplicity).
Post hoc analyses
Additional exploratory analyses included investigations of: whether there was a responder subpopulation that could be defined by baseline/demographic characteristics; the effect of AZD9668 during the course of an exacerbation; biomarker patterns in ‘chronic bronchitic’ subgroups (BCSS cough and sputum each ≥2; also, BCSS cough and sputum ≥2 plus Bronkotest® colour ≥2) (see Supplementary Material for further information on post hoc analyses).
Ethical aspects
The study was conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonisation/Good Clinical Practice guidelines, applicable regulatory requirements, and AstraZeneca's policy on bioethics. All patients provided written informed consent. The final study protocol was approved by Independent Ethics Committees/Institutional Review Boards, and an independent Safety Review Committee reviewed accumulated blinded safety data on an ongoing basis during the study.
Results
Patients
Eight hundred and thirty-eight patients, enrolled between July 2009 and August 2010, were randomised and received study medication (). Baseline demography and clinical characteristics were well balanced between the treatment groups (). The mean compliance with study treatment was 92–94% and with maintenance tiotropium was 91–92%. There were no differences between the groups for COPD medications received before randomisation and during the randomised treatment period (see Supplementary Material).
Figure 2. Patient disposition during the study.
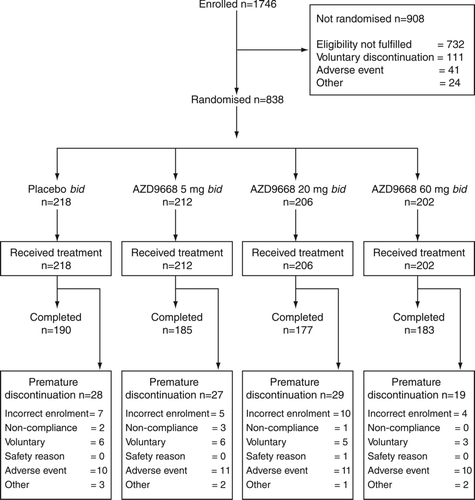
Table 1. Baseline demography and clinical characteristics (safety analysis set)
Efficacy
Lung function variables: Mean pre-bronchodilator FEV1 at baseline was balanced across the treatment groups (1.47–1.51 L). In the AZD9668 60 mg bid group at the end of treatment, the change in mean pre-bronchodilator FEV1 compared with placebo was 0.00 L (95% CI: −0.05, 0.04; p = 0.873) (). (In accordance with the hierarchical decreasing procedure described in the statistical methodology, as statistical significance was not met at the highest dose level, no analyses were carried out to compare the lower doses.)
Figure 3. Comparison of pre-bronchodilator lung function and daily lung function FEV1 and PEF between AZD9668 and placebo. LSM differences and 95% CIs between AZD9668 and placebo for (A) pre-bronchodilator lung function at Week 12, (B) daily lung function FEV1 (morning and evening) (mean of the last 6 weeks on treatment) and (C) daily lung function PEF (morning and evening) (mean of the last 6 weeks on treatment). The ANCOVA model included country and baseline as covariates. If the CI contains 0, there is no difference between AZD9668 and placebo.
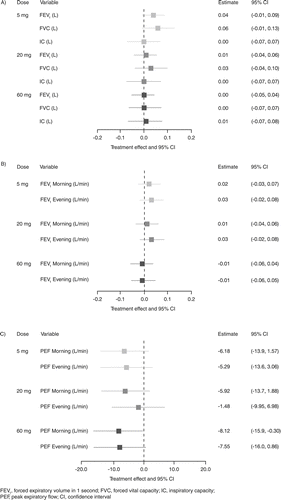
Mean post-bronchodilator FEV1 at baseline was also balanced across the groups (1.64–1.69 L). In the AZD9668 60 mg bid group at the end of treatment, the change in mean post-bronchodilator FEV1 compared with the placebo was 0.01 L (95% CI: −0.04, 0.06; p = 0.667).
Mean pre-bronchodilator FVC and IC at baseline were 3.01–3.08 L and 2.14–2.22 L, respectively. In the AZD9668 60 mg bid group at the end of treatment, the change in mean values for pre-bronchodilator FVC and IC were 0.00 L (95% CI: −0.07, 0.07; p = 0.894) and 0.01 L (95% CI: −0.07, 0.08; p = 0.881), respectively (). Post-bronchodilator FVC and IC were also unchanged at the end of treatment.
FEV1 measured at home (morning and evening) at baseline was well balanced across the groups and during the last 6 weeks of treatment there was no difference between AZD9668 60 mg bid and placebo in change from baseline in mean FEV1: the change in mean FEV1 (morning and evening) compared with placebo was −0.01 L/min (95% CI: −0.06, 0.04; p = 0.620) and −0.01 L/min (95% CI: −0.06, 0.05; p = 0.814), respectively ().
There was a significant difference for mean PEF measured at home for AZD9668 60 mg bid at the end of treatment in favour of placebo: the change in mean PEF (morning and evening) compared with placebo was −8.12 L/min (95% CI: −15.9, −0.30; p = 0.042) and −7.55 L/min (95% CI: −16.0, 0.86; p = 0.079), respectively ().
Signs and symptoms. The mean (standard deviation) BCSS diary card scores at baseline were well balanced across the groups (4.5 [1.8], 4.9 [2.0], 4.7 [1.5] and 4.8 [1.8] in the placebo, AZD9668 5, 20 and 60 mg bid groups, respectively). During the last 6 weeks of treatment there were no differences between AZD9668 60 mg bid and placebo in the change from baseline in diary card scores (–0.4 [1.6], −0.6 [1.4], −0.6 [1.5] and −0.4 [1.5] in the placebo, AZD9668 5, 20 and 60 mg bid groups, respectively). Similarly, there were no differences between groups in changes in EXACT scores.
Baseline and change from baseline data for other measures of signs and symptoms are summarised in Supplementary Material . Sputum colour at baseline was well balanced across the groups (2.13–2.34). At the end of treatment there was no difference between the AZD9668 60 mg bid group and the placebo group for the change from baseline in mean sputum colour.
Reliever medication usage during the last 6 weeks of treatment and exercise capacity, as measured by the 6-minute walk test, at the end of treatment was largely balanced across the groups at baseline and did not differ between the AZD9688 60 mg bid group and placebo group.
The SGRQ for COPD patients (SGRQ-C) and FPI-SF (completed by 178 patients in English speaking countries) scores at baseline were balanced across the groups. The SGRQ-C scores at the end of treatment were improved in all groups. An analysis of the overall score for SGRQ-C at the end of treatment showed improvements in favour of placebo, with LSM differences of 3.15 (95% CI: 0.46, 5.85; p = 0.022), 3.14 (95% CI: 0.40, 5.88; p = 0.025) and 1.58 (95% CI: −1.13, 4.29; p = 0.251) over the AZD9668 5, 20 and 60 mg bid groups, respectively; the pattern seen for the symptom, activity and impact scores was similar to the overall score. At the end of treatment, FPI-SF mean scores in each group were unchanged from baseline values; there were no differences observed in FPI-SF scores for the comparison between the AZD9668 60 mg bid group and the placebo group.
The total number of patients with 1 or more exacerbations was 29 (13%), 29 (14%), 28 (14%) and 34 (17%) in the placebo, AZD9668 5, 20 and 60 mg bid groups, respectively. The duration of exacerbation across the groups was broadly similar at 7.8–10.0 days. An analysis of time to first clinic defined exacerbation showed no statistically significant differences between AZD9668 and placebo (5 mg bid, p = 0.873; 20 mg bid, p = 0.910; 60 mg bid, p = 0.318). Amongst all study subjects, 21.5% who were on combined therapy with ICS/LABA prior to study entry had exacerbations compared with 11.9%, who were on neither.
Exploratory endpoints: biomarker data
An analysis of blood inflammatory markers for TNF-α, IL-8 and IL-6 showed no difference at the end of treatment between each AZD9668 treatment group versus placebo (Supplementary Material Table 2), with the end of treatment LSM ratios between AZD9668 and placebo at or close to 1. Geometric mean desmosine (free) normalised for creatinine levels at baseline were 1.99, 1.98, 1.91 and 1.99 nmol/mmol in the placebo, AZD9668 5, 20 and 60 mg bid groups, respectively. At the end of treatment, the LSM values were 2.01, 2.10, 2.06 and 2.16 nmol/mmol, respectively (Supplementary Material Table 2). ANCOVA for free desmosine (normalised for) creatinine showed no significant differences between AZD9668 and placebo at the end of treatment (5 mg bid, p = 0.456; 20 mg bid, p = 0.704; 60 mg bid, p = 0.241).
Pharmacokinetics
The Day 84 pre-dose geometric mean concentrations were 27.0, 83.8 and 262.7 nM for AZD9668 5, 20 and 60 mg bid, respectively, showing an approximately dose-proportional increase. Plasma geometric mean concentrations at the estimated peak concentration (0.5–4 hours post-dose) were 73.4, 252.5 and 621.7 nM, respectively. The plasma concentrations at Day 28 were similar.
Post hoc analyses
Post hoc exploratory analyses found no significant (p > 0.1 for all values) interactions between baseline characteristics and change in FEV1, SGRQ-C and BCSS. There were no significant (p > 0.1 for all values) interactions between exacerbation severity and PEF/home FEV1, BCSS or reliever medication or between ‘chronic bronchitic’ subsets (symptom subgroups set up as surrogates for a bronchitic phenotype) and biomarkers.
Safety
The numbers of patients with AEs (84 [39%], 78 [37%], 66 [32%] and 67 [33%] for placebo and AZD9668 5, 20 and 60 mg bid groups, respectively) and with AEs leading to discontinuation (10 [5%], 11 [5%], 12 [6%] and 10 [5%] for placebo and AZD9668 5, 20 and 60 mg bid groups, respectively) were similar in each treatment group. Although the number of patients with serious AEs (SAEs) was higher in both the AZD9668 20 and 60 mg bid groups versus the placebo and AZD9668 5 mg groups, there was no clear evidence of dose dependency. The most frequently reported SAE was COPD (exacerbation) with 4 (2%), 1 (<1%), 7 (3%) and 7 (3%) patients in the placebo, AZD9668 5, 20 and 60 mg bid groups, respectively.
With the exception of pneumonia which occurred in 2 patients in the 20 mg group, no other SAE occurred in >1 patient in a treatment group. One patient experienced an AE with fatal outcome during the study (placebo group: pulmonary thrombosis/embolism). Few AEs were reported at an incidence of >2% in any treatment group and included nasopharyngitis, COPD, upper respiratory tract infection, headache and dry mouth. The most common AE in each group was nasopharyngitis. There was no apparent evidence of any dose dependency in tolerability. Few patients experienced severe AEs with 2 (1%), 7 (3%), 8 (4%) and 5 (2%) patients in the placebo, AZD9668 5, 20 and 60 mg bid groups, respectively.
There was no evidence of dose dependency in haematology or clinical chemistry values over time and no apparent differences between AZD9668 compared with placebo. A small but higher number of patients in the AZD9668 groups had clinically significant elevations in transaminase levels versus placebo (elevation of alanine transaminase [ALT] to ≥3x upper limit of normal [ULN] occurred in 1 [<1%], 4 [2%], 1 [<1%] and 4 [2%] in the placebo, AZD9668 5, 20 and 60 mg bid groups, respectively, and elevation of aspartate transaminase [AST] to ≥3xULN occurred in 2 [1%], 5 [2%], 1 [<1%] and 3 [1%] patients in the placebo, AZD9668 5, 20 and 60 mg bid groups, respectively). There were confounding factors (e.g. alcohol intake) in some but not all cases and thus a definitive relationship to study drug could not be ruled out.
Discussion
Treatment for up to 3 months with AZD9668 did not improve lung function, respiratory signs and symptoms or QoL when added to a long-acting muscarinic antagonist (LAMA) in patients with symptomatic COPD with a history of at least one exacerbation in the year before. There was no evidence of any effect on serum inflammatory markers or on urinary desmosine, a marker of elastin degradation. Also, there was no consistent evidence from the post hoc analyses of a beneficial effect of AZD9668 in any specific patient subgroup. There was a lack of change in sputum colour in this study; however, baseline sputum colour scores were low in this study, which would have limited the potential for improvements.
AZD9668, in combination with a LAMA, was generally well tolerated. Across the AZD9668 5, 20 and 60 mg bid treatment groups, there was no evidence of any dose-dependent increase in AEs, SAEs or AEs leading to discontinuation and no differences from placebo. A few patients on AZD9668 developed elevated transaminase values, for which relationship to study drug could not be definitely excluded. The highest transaminase elevations occurred at AZD9668 60 mg but there was no definitive evidence of a dose relationship.
The failure of this agent to show any effects on usual endpoints and markers of COPD was seen in a previous study by Luisetti et al, who also investigated an elastase inhibitor, MR889, administered over a period of 4 weeks to COPD patients (Citation9). In this double-blind, randomised, placebo-controlled clinical trial of 60 COPD patients, MR889 did not modify biochemical markers of lung destruction in unselected COPD patients. However, in contrast to our study, the level of urinary desmosine dropped significantly (p = 0.004) with respect to pre-treatment values in a subset of treated patients with a short disease duration.
The lack of efficacy of AZD9668 in this study could be due to a number of reasons. For example, it may be reflective of the ability of AZD9668 to show efficacy in only a specific subset of patients as reported by Luisetti et al, (Citation9). Alternatively, the physical properties of NE in this population of patients may have prevented the efficacy of AZD9668. Travis et al, have shown bound proteinases to exhibit an increased resistance to inhibition by proteinase inhibitors (Citation10). The study was designed to assess questions of relatively short-term clinical relevance (lung function and symptoms). This may have contributed to the lack of AZD9668 efficacy on exploratory endpoints such as desmosine levels.
Decreasing levels of desmosine would have been indicative of a decrease in elastin degradation and thus a direct effect of AZD9668 on disease modification. Although it is likely that studies of longer than 3 months would be required to see the full effect of a potential disease-modifying agent in this chronic disease, long-term trials are difficult to perform in patients with COPD: patients are not easy to recruit and many patients fail to complete the studies, with significantly more patients withdrawing from the placebo arms than the active treatment arms (Citation11,12) leading to significant bias (Citation13). In this 3-month study on patients maintained on tiotropium, this effect was less likely to have played a role as demonstrated by relatively similar withdrawal rates across groups.
The doses of AZD9668 used in this study were based on preclinical (Citation7) and subsequent Phase I/IIa studies (Gunawardena et al, submitted), which showed that 70 mg bid was the maximum permitted dose based on preclinical toxicity data. At this dose, AZD9668 levels were approximately 60-fold higher than the half maximal inhibitory concentration in vitro (Citation7) (Gunawardena et al, submitted). A dose of AZD9668 60 mg bid has also shown preliminary positive findings in other Phase II studies.
A significant improvement in FEV1 versus placebo was found after 4 weeks of treatment in patients with idiopathic bronchiectasis, although no significant difference in the primary endpoint of sputum neutrophil counts was observed (Citation14). In a second study in patients with cystic fibrosis, although AZD9668 demonstrated no effect on lung function, neutrophil counts or NE activity, it did reduce inflammatory sputum biomarkers and elastin degradation markers (Citation15). However, bronchiectasis and cystic fibrosis are phenotypically distinct from COPD. The increased burden of NE and other inflammatory mediators in these diseases may have contributed to the positive results seen with AZD9668 treatment and may not be reproducible in a disease of lower inflammatory burden such as COPD.
It is possible that in this study, the concentration of AZD9668 may have been inadequate to inhibit tissue NE. However, in the current study, the geometric mean plasma concentration of AZD9668 at 0.5–4 hours following a dose of 60 mg bid (622 nM) was consistent with that in a Phase IIa study in patients with COPD (Gunawardena et al, submitted). Another explanation for the lack of efficacy in this study is that AZD9668 did not reach its site of action. This explanation is countered by the results of the same Phase IIa study, which detected AZD9668 in the sputum of COPD patients at clinically relevant concentrations (Gunawardena et al, submitted). One other possibility is that AZD9668 is inactivated when it enters the lung. However, it is not known whether the innate mechanisms involved in the inactivation of endogenous inhibitors of NE (Citation16,17) would also affect AZD9668.
Although currently available bronchodilator-based therapies ameliorate the symptoms of COPD, a disease-modifying effect, i.e., a decrease in the rate of disease progression (Citation18) has been more difficult to demonstrate. A large-scale trial including nearly 6000 patients with moderate-to-severe COPD (Understanding Potential Long-term Impacts on Function with Tiotropium [UPLIFT]) aimed to determine whether tiotropium decreased the rate of pre- and post-bronchodilator FEV1 over a 4-year period (Citation12). Although treatment with tiotropium improved lung function, QoL and exacerbations it did not significantly reduce the rate of decline in FEV1.
As we enter a new era of research, beyond LAMAs, LABAs and ICSs, there has been increasing interest in the use of biomarkers to understand and monitor airway inflammation in patients with respiratory tract disorders, such as COPD. Lung lavage or induced sputum studies could act as an interim step when transitioning from Phase IIa to Phase IIb studies, if biomarkers could be found that reflect inflammatory changes in the airways over a short period of time.
However, an improved understanding of the changes in biomarker levels during the inflammatory process is needed. Currently, no biomarkers have been shown to be associated with a reduction in inflammation or disease severity in patients with COPD (Citation19) and a meta-analysis failed to find a significant association between serum CRP or serum TNF levels and COPD disease stages (Citation20). Recently, Carter et al, demonstrated that the fibrinogen cleavage product, Aα-Val360, is a specific marker of NE activity in vivo and hence is a potential biomarker of disease activity and progression in subjects with elastase-dependent COPD (Citation21).
Given the current lack of biomarkers that clearly reflect inflammatory changes in the airways over a short period of time, and the potential ability of anti-inflammatory agents to modify static lung volumes, it may have been beneficial to include the latter as well as Aα-Val360 as study endpoints assessed in the current study and certainly this is a learning for the design of future trials investigating disease-modifying agents. Nevertheless, it is clear that new study concepts and novel endpoints are required for the study of purely anti-inflammatory drugs, particularly those targeted for long-term effects. For example, there is evidence that AZD9668 inhibits NE in healthy volunteers (Gunawardena et al, submitted) but no evidence for NE inhibition at site of action in COPD patients. Definitive evidence of NE inhibition in COPD patients might help interpret the results of future trials of disease-modifying agents. Furthermore, there may be a need to better phenotype the heterogeneous COPD patient population (Citation22) to define those patients who are most likely to respond to more targeted therapies.
In conclusion, the current study robustly addresses questions related to relatively short-term efficacy of evaluated doses of AZD9668 in patients with symptomatic COPD maintained on tiotropium. Although preclinical and preliminary Phase I/II data showed promising results in patients with chronic respiratory tract disorders, this did not translate into significant changes in lung function, signs or symptoms in this 3-month trial in patients with COPD. The results of this study are corroborated by the negative results of a similar Phase IIb dose-finding study (NCT01023516) that was conducted in parallel to assess the efficacy and safety of AZD9668 administered to patients with COPD receiving maintenance budesonide/formoterol (Kuna et al, submitted). Taken together, the results of both these studies seem to challenge the role of NE in COPD pathogenesis; however, it is not clear whether the results obtained in this study reflect an overall lack of efficacy of AZD9668 or whether they reflect limitations in the dose selection or endpoint assessments. Nevertheless, this is a new area of research in the treatment of chronic inflammatory lung disease and the data generated contribute important information on study design in this field.
Declaration of Interest Statement
This study was funded by AstraZeneca. Medical writing assistance was provided by Dr Annette Smith from Complete Medical Communications, funded by AstraZeneca.
Claus Vogelmeier has given presentations at symposia sponsored by AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Janssen-Cilag, Novartis, Nycomed, Pfizer and Talecris, and has received consultancy fees from AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Janssen-Cilag, Mundipharma, Novartis, Nycomed and Talecris. Teresita Aquino has no conflicts of interest. Christopher O'Brien is an AstraZeneca employee and stockholder. John Perrett is a former AstraZeneca employee. Kulasiri Gunawardena is a former AstraZeneca employee and current Novartis employee.
Authorship contributions: Claus Vogelmeier - principal investigator, Teresita O Aquino - planning and interpretation of biomarker data, Christopher O'Brien - medical officer responsible for study co-ordination and implementation, as well as data interpretation, John Perrett - data analysis, presentation and interpretation and Kulasiri Gunawardena - study design, medical monitoring and data interpretation. All authors developed, reviewed and commented on draft versions and approved the final manuscript.
Principal investigators
Australia - Matthew Peters, Paul Seale, Maureen McKeirnan, Mark Holmes, Huw Davies, Peter Frith, Philip J Thompson, Abe Rubinfeld, Martin Phillips, Steven Lindstrom. Canada - Denis O'Donnell, Tharwat Fera, Richard Tytus, Emad Amer, Giuseppe Mazza, Jacques Bouchard, Jacques Hebert, Dennis O'Keefe, A William Booth, Arun Nayar, Qaiser Rizvi, Allan Kelly, Andy Lam, Darcy Marciniuk, Guy Tellier, Andre Frechette, Anthony D'Urzo. Japan - Akinori Takeda, Noriharu Shijubo, Shigeru Komatsu, Sekiya Koyama, Yoshikazu Inoue, Motokazu Kato, Yuji Nakatani, Eishin Shimizu, Hiroyuki Akiyama, Tsutomu Iwasa, Makoto Hagiwara, Soichiro Hozawa, Michikazu Terada, Shuichi Yano, Masahiro Abe, Fumitaka Ogushi, Shinichi Tanaka, Naoki Hagimoto, Mitsuru Masuda, Takao Tochigi, Hiroyuki Taniguchi, Yasuhiro Kondo, Mayumi Eiro, Hiroshi Koto, Naoki Miyao, Hiroki Ninomiya, Naruhiko Sugihara, Arihiro Kiyosue, Haruhisa Kuroda, Kiyotaka Nakajima. Germany - Claus Franz Vogelmeier, Karin Förster, Anneliese Linnhoff, Hermann Trauth, Lutz Von Versen, Jörg Winkler, Stephan Molitor, Reiner Laumen, Gerhard Hoheisel, Ruth Nischik. Philippines - Teresita Aquino, Tito Atienza, Ronnie Samoro. Poland - Piotr Nalepa, Cezary Rybacki, Agata Kot, Beata Asankowicz-Bargiel, Wojciech Skucha, Zbigniew Bajor, Krzysztof Wytrychowski, Wiesaw Andrzejewski, Krzysztof Lis, Lucyna Dymek, Ewa Springer, Krzysztof Buczyko, Danuta Madra-Rogacka. Republic of Korea - Kwan Hyoung Kim, Jae Jeong Shim, Kwang Ho In, Choon-Sik Park, Suk-Joong Yong, Kwan-Ho Lee, Ki-Suck Jung, Ho Joo Yoon, Sang Haak Lee. Russian Federation - Alla Tsoy, Y Popova, A G Chuchalin, S Mikhailov, Alexander Emelyanov, Mikhail Ilkovich, A Galustyan, Olga Smolenskaya, T Martynenko, A Krivosheev, R Fassakhov, Alexandr Vizel, Mikhail Kotelnikov, V Podzolkov. Slovakia - Yveta Kubikova, Maria Paluchova, Helena Horvathova, Katarina Arpasova, Helena Lescisinova, Svetlana Kurthova, Martin Sisan, Maria Pobehova, Luboslava Frajtova, Jozef Komada, Erika Pribulova, Helena Hukelova, Denisa Kavkova. Taiwan - Chi-Huei Chiang, Chong-Jen Yu, Liang-Wen Hang, Chao-Chien Wu, Ming-Shyan Huang, Shih-Lung Cheng, Gwan-Han Shen, Yu-Chih Liu. Ukraine - Yuriy Feshchenko, Liudmyla Yashina, Volodymyr Gavrysyuk, Oleksandr Dzyublyk, Liliia Romaniuk, Tatyana Pertseva, Volodymyr Yefimov, Viktor Blazhko, Yuriy Kolchin, Mykola Ostrovskyy, Orest Abrahamovych. USA - John Southard, Michael Desantis, David Laman, James Pearle, Gregory Feldman, Richard Martinez, David Erb, Robert Buynak, Krishna Pudi, Ritsu Kuno.
Acknowledgments
We thank the investigators and patients for their participation in this study.
References
- From the Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2010. Available from: http://www.goldcopd.org/ (accessed 22 December, 2011).
- Quint JK, Wedzicha JA. The neutrophil in chronic obstructive pulmonary disease. J Allergy Clin Immunol 2007; 119:1065–1071.
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, Pare PD. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004; 350:2645–2653.
- Cowburn AS, Condliffe AM, Farahi N, Summers C, Chilvers ER. Advances in neutrophil biology: clinical implications. Chest 2008; 134:606–612.
- Kawabata K, Suzuki M, Sugitani M, Imaki K, Toda M, Miyamoto T. ONO-5046, a novel inhibitor of human neutrophil elastase. Biochem Biophys Res Commun 1991; 177:814–820.
- Lucas SD, Costa E, Guedes RC, Moreira R. Targeting COPD: Advances on low-molecular-weight inhibitors of human neutrophil elastase. Med Res Rev 2011; [Epub ahead of print].
- Stevens T, Ekholm K, Granse M, Lindahl M, Kozma V, Jungar C, Ottosson T, Falk-Hakansson H, Churg A, Wright JL, Lal H, Sanfridson A. AZD9668: pharmacological characterisation of a novel oral inhibitor of neutrophil elastase. J Pharmacol Exp Ther 2011; 339:313–320.
- Gunawardena K, Gullstrand H, Perrett J. Safety, tolerability and pharmacokinetics of AZD9668, an oral neutrophil elastase inhibitor, in healthy subjects and patients with COPD. Eur Respir J 2010; 203s.
- Luisetti M, Sturani C, Sella D, Madonini E, Galavotti V, Bruno G, Peona V, Kucich U, Dagnino G, Rosenbloom J, Starcher B, Grassi C. MR889, a neutrophil elastase inhibitor, in patients with chronic obstructive pulmonary disease: a double-blind, randomized, placebo-controlled clinical trial. Eur Respir J 1996; 9:1482–1486.
- Bangalore N, Travis J. Comparison of properties of membrane bound versus soluble forms of human leukocytic elastase and cathepsin G. Biol Chem Hoppe Seyler 1994; 375:659–666.
- Celli BR, Thomas NE, Anderson JA, Ferguson GT, Jenkins CR, Jones PW, Vestbo J, Knobil K, Yates JC, Calverley PM. Effect of pharmacotherapy on rate of decline of lung function in chronic obstructive pulmonary disease: results from the TORCH study. Am J Respir Crit Care Med 2008; 178:332–338.
- Tashkin DP, Celli B, Senn S, Burkhart D, Kesten S, Menjoge S, Decramer M. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008; 359:1543–1554.
- Vestbo J, Anderson JA, Calverley PM, Celli B, Ferguson GT, Jenkins C, Yates JC, Jones PW. Bias due to withdrawal in long-term randomised trials in COPD: evidence from the TORCH study. Clin Respir J 2011; 5:44–49.
- Stockley RA, Snell N, Perrett J, Gunawardena K. Efficacy and safety of AZD9668, an oral neutrophil elastase inhibitor, in idiopathic bronchiectasis. Eur Respir J 2010; 38s.
- Elborn JS, Perrett J, Forsman-Semb K, Marks-Konczalik J, Gunawardena K, Entwistle N. Effect of the oral neutrophil elastase inhibitor, AZD9668, in patients with cystic fibrosis. Abstract presented at the 107th Annual Conference of the American Thoracic Society, Denver, Colorado; May 13–18, 2011.
- Johnson D, Travis J. The oxidative inactivation of human alpha-1-proteinase inhibitor. Further evidence for methionine at the reactive center. J Biol Chem 1979; 254:4022–4026.
- Zeiher BG, Matsuoka S, Kawabata K, Repine JE. Neutrophil elastase and acute lung injury: prospects for sivelestat and other neutrophil elastase inhibitors as therapeutics. Crit Care Med 2002; 30:S281–S287.
- Halpin DM, Tashkin DP. Defining disease modification in chronic obstructive pulmonary disease. COPD 2009; 6:211–225.
- Vestbo J, Rennard S. Chronic obstructive pulmonary disease biomarker(s) for disease activity needed–urgently. Am J Respir Crit Care Med 2010; 182:863–864.
- Franciosi LG, Page CP, Celli BR, Cazzola M, Walker MJ, Danhof M, Rabe KF, la Pasqua OE. Markers of disease severity in chronic obstructive pulmonary disease. Pulm Pharmacol Ther 2006; 19:189–199.
- Carter RI, Mumford RA, Treonze KM, Finke PE, Davies P, Si Q, Humes JL, Dirksen A, Piitulainen E, Ahmad A, Stockley RA. The fibrinogen cleavage product Aa-VAL360, a specific marker of neutrophil elastase activity in vivo. Thorax 2011; 66:686–691.
- Han MK, Agusti A, Calverley PM, Celli BR, Criner G, Curtis JL, Fabbri LM, Goldin JG, Jones PW, MacNee W, Make BJ, Rabe KF, Rennard SI, Sciurba FC, Silverman EK, Vestbo J, Washko GR, Wouters EF, Martinez FJ. Chronic obstructive pulmonary disease phenotypes: the future of COPD. Am J Respir Crit Care Med 2010; 182:598–604.
- From the Global Strategy for Asthma Management and Prevention, Global Initiative for Asthma (GINA) 2010. Available from: http://www.ginasthma.org/ (accessed 22 December, 2011).