Abstract
Introduction: Animal studies demonstrate the importance of the E3 ubiquitin ligases, Muscle RING-Finger Protein 1 (MuRF-1) and atrogin-1, in muscle protein degradation during acute muscle atrophy. Small clinical studies suggest MuRF-1 and atrogin-1 expression in the quadriceps muscle is also increased in stable patients with Chronic Obstructive Pulmonary Disease compared to controls. However, it remains unclear whether these ligases have a role in maintaining a muscle-wasted state in COPD patients. Methods: 32 stable COPD patients (16 with a low fat-free mass index (FFMI), 16 with a normal FFMI) and 15 controls underwent lung function and quadriceps strength tests and a percutaneous quadriceps biopsy. Quadriceps MuRF-1 and atrogin-1 protein were quantified with western blotting. Quadriceps fiber cross-sectional area (CSA) and fiber proportions were determined by immunohistochemistry on muscle sections. MuRF-1 and atrogin-1 levels were compared between COPD patients with and without a low FFMI, and between patients and controls, and correlations between MuRF-1 and atrogin-1 levels and quadriceps fiber CSA in the patients were investigated. Results: Atrogin-1 protein levels were lower in patients than controls, but similar in patients with a low and normal FFMI. MuRF-1 levels did not differ between any groups. MuRF-1 and atrogin-1 levels were not associated with quadriceps fiber CSA or quadriceps strength in patients. Conclusions: Chronic upregulation of ubiquitin ligases was not evident in the quadriceps muscle of stable COPD patients with a low muscle mass. This does not exclude the possibility of transient increases in ubiquitin ligases during acute catabolic episodes.
Introduction
Loss of muscle bulk, particularly in the lower limb, is a common and important complication of COPD (Citation1) predicting a poor outcome independent of the severity of lung disease (Citation2, 3). This has led to interest in the molecular mechanisms underlying muscle atrophy in COPD.
The ubiquitin-proteasome pathway is a key final pathway of muscle protein breakdown. Enzymes involved in this pathway, in particular Muscle RING-Finger Protein-1 (MuRF-1) and atrogin-1, mediate acute muscle atrophy in experimental animal and cell models (Citation4). MuRF-1 and atrogin-1 expression are induced by nuclear factor-kappa B (Citation5) and P38 mitogen-activated protein kinase activation (Citation6) respectively; activation of these pathways may or may not occur in the muscle of patients with COPD (Citation7–11).
In one study of 12 stable COPD patients, increased MuRF-1 and atrogin-1 mRNA levels were found in the quadriceps of patients compared to controls but there were no differences in either mRNA between patients with low muscle mass and patients with normal muscle mass (Citation12), no differences in atrogin-1 protein between patients with low muscle mass and patients with normal muscle mass, and MURF-1 protein was not measured. Another study reported increased atrogin-1 mRNA and increased Nedd4 protein (another ubiquitin ligase), but not increased MuRF-1 protein, in the quadriceps of nine patients with muscle atrophy compared to nine controls. However in that study, the relationship between these proteins and muscle mass or quadriceps function were not explored (Citation13).
There is, therefore, insufficient data comparing MuRF-1 and atrogin-1 protein levels in COPD patients with and without muscle atrophy to clarify whether these proteins are likely to have a role in determining muscle mass in stable COPD. Therefore, we compared protein expression of MuRF-1 and atrogin-1 in quadriceps samples from 32 COPD patients (16 with muscle atrophy as defined by a low global fat-free mass index using the Dutch criteria (Citation14), 16 without muscle atrophy) and 15 healthy age-matched controls, and examined relationships with muscle mass and quadriceps fiber size. Our conclusions were further supported by analysis of MuRF-1 and atrogin-1 mRNA in another, overlapping cohort of COPD patients and controls (details in supplement).
Methods
Ethical approval
Study numbers 06/Q0404/35 and 06/Q0410/54 were approved by the Royal Brompton & Harefield NHS Trust and Ealing and West London Mental Health Trust Research Ethics Committees and all participants gave written, informed consent.
Participants
First, 32 GOLD(Citation15) Stage II to IV COPD patients were enrolled from respiratory clinics and 15 healthy controls were recruited by advertisement. A diagnosis of heart, renal or liver failure, a systemic inflammatory, metabolic or neuromuscular disorder or a severe exacerbation (ie requiring antibiotics, oral steroids, or hospitalisation) within the previous 4 weeks or warfarin therapy (bleeding risk from biopsy) were exclusion criteria.
Physiological measurements and quadriceps biopsy
Post-bronchodilator spirometry (Citation16), lung volumes (by plethysmography) (Citation17), diffusion capacity (Citation18) and arterialized capillary earlobe blood gas tensions were measured. Physical activity was measured using a tri-axial accelerometer (Dynaport ADL; McRoberts BV, the Netherlands) for 12 hours/day for 2 days as validated by Pitta et al (Citation19). Fat-free mass index (FFMI) was calculated from bioelectrical impedance measurements (Bodystat 1500, UK) taken after participants had rested supine for 20 minutes, using a disease-specific regression equation (Citation20).
Patients were classified as having a low or normal FFMI using the Dutch criteria [15 and 16 kg/m2 cut-off for females and males respectively (Citation14)]. Quadriceps strength (dominant leg) was assessed by supine isometric maximal voluntary contraction (MVC) (Citation21) and unpotentiated twitch quadriceps force (TwQ) (Citation22). Exercise performance was assessed with a 6-minute walking test (6MW) performed according to ATS guidelines (Citation23) and symptom-limited incremental cycle ergometry with metabolic testing as described previously (Citation24). On a separate occasion, percutaneous needle biopsy of the vastus lateralis was performed using the Bergstrom technique and samples stored at –80°C (Citation25) (see supplement).
Quantification of MURF-1 and atrogin-1 protein in quadriceps muscle
Protein levels were determined by Western blot using anti-MuRF1, anti-atrogin-1 and anti-alpha tubulin antibodies (see supplement).
Measurement of quadriceps muscle fiber cross-sectional area (CSA) from biopsy
Immunohistochemistry was performed on 10 μm transverse muscle sections using anti-type I myosin, anti-type IIa myosin and anti-laminin antibodies to determine type I, type I/IIa, IIa, IIx proportions and median CSA of each fiber type (Citation26, 27) from a minimum of 100 fibers per subject (see supplement). The patients in this MS (in whom ubiquitin ligases) were measured were also participants in a larger study describing the heterogeneity of quadriceps biopsy appearances in COPD (Citation28).
Statistical analysis
Sample size calculation
Using the MuRF-1 protein data from COPD patients and controls in the publication of Plant et al. (Citation13), a sample size of 11 was required in both the COPD and control groups to achieve 90% power. A similar calculation using the atrogin-1 protein data from the paper of Doucet et al. (Citation12) suggested a sample size of 7. Atrogin-1 protein and MuRF-1 mRNA expression were not included in Doucet's paper for the COPD patients with and without muscle wasting, so we were unable to calculate the sample size required for the low and normal FFMI groups.
Data analysis
Normally and non-normally distributed data are presented as mean (standard deviation) and median (25th percentile, 75th percentile) respectively, and group differences assessed with a t-test and the Mann–Whitney U-test respectively. Differences between categorical variables were tested with Fisher's exact test. Spearman's rank correlation coefficient was calculated to assess correlations between MURF-1 and atrogin-1 levels and FFMI and quadriceps fiber CSA. A conventional 2-tailed p-value of ≤ 0.05 was used to define statistical significance.
Results
The clinical and physiological characteristics of the COPD patients and controls are shown in Tables and . As expected, the COPD patients had significantly poorer lung function, arterial oxygen tensions, FFMI, quadriceps strength, exercise performance and physical activity levels than the controls (). Patients with a low FFMI had a significantly lower TLCO, but not lower FEV1, than patients with a normal FFMI, and as expected quadriceps strength was lower in the patients with a low FFMI compared to patients with a normal FFMI. Patients with a low FFMI also had a lower BMI and greater pack-year smoking history than patients with a normal FFMI but otherwise the low and normal FFMI groups did not differ significantly, including the proportion of patients on long-term oral glucocorticoids ().
Table 1. Clinical characteristics of COPD patients and controls
Table 2. Quadriceps muscle fiber cross-sectional areas in COPD patients and controls
Patients had significantly reduced glycolytic type IIx fiber CSA [2490(1830,3240) μm2 vs 4790(2930,6190)μm2, p = 0.003, ], a reduced proportion of oxidative type I fibers [28(19)% v 58(14)% p < 0.0001], and increased proportions of intermediate oxidative/glycolytic type IIa [62(17)% vs 37(14)% p < 0.0001] and glycolytic type IIx fibers compared to controls [4(1,8)% vs 0(0,4)%% p = 0.009]. Quadriceps muscle fiber CSA was not significantly different between patients with and without a low FFMI (type I fibers 4650(3630,7230) μm2 vs 5070(3670,5720) μm2, p = 1.00, type IIa fibers 3520(2220,4850) μm2 vs 4130(3670,4750) μm2, p = 0.26 and type IIx fibers 2550(1650,3370) μm2 vs 2490(1930,3920) μm2, p = 0.59, ) and type I and II fiber proportions did differ between these groups.
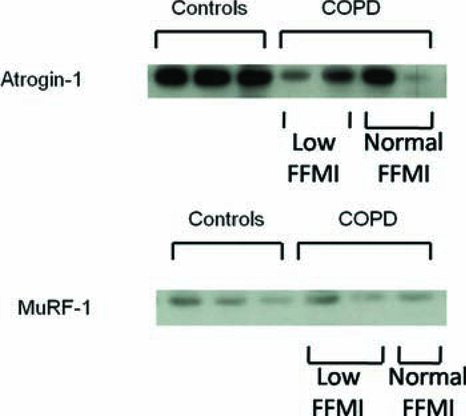
Quadriceps MuRF-1 and atrogin-1 protein levels in the patient and control groups are displayed in and representative images of the Western blots shown in . Atrogin-1 protein levels were significantly lower in COPD patients than controls [0.64(0.31,1.24)AU vs 1.41(0.68,2.06)AU, p = 0.03, ]. However, atrogin-1 protein levels were not significantly different between COPD patients with and without a low FFMI [0.73(0.34,1.61)AU vs 0.74(0.12,1.34)AU, p = 0.46, ], with a small subset of patients in both the normal and low muscle mass groups having extremely high and low values compared to controls.
Figure 1. Box plots of atrogin-1 and MuRF-1 protein levels in COPD patients with and without a low FFMI and healthy age-matched controls. Atrogin-1 protein levels in quadriceps muscle were lower in COPD patients than controls [0.64(0.31,1.24)AU vs 1.41(0.68,2.06)AU, p = 0.03, A] but not significantly different in COPD patients with a reduced FFMI compared to patients with a normal [0.73(0.34,1.61)AU vs 0.74(0.12,1.34)AU, p = 0.46, A]. There was a subset of COPD patients with relatively high protein levels of these mediators compared to the controls but these patients were not confined to the low FFMI group. MuRF-1 protein levels were not significantly different in quadriceps muscle from COPD patients compared to controls [0.56(0.45,1.30)AU vs 0.92(0.74,1.50)AU, p = 0.12, B], nor between COPD patients with and without a low FFMI [0.55(0.42,0.92)AU vs 0.59(0.54,1.62)AU, p = 0.35, B].
![Figure 1. Box plots of atrogin-1 and MuRF-1 protein levels in COPD patients with and without a low FFMI and healthy age-matched controls. Atrogin-1 protein levels in quadriceps muscle were lower in COPD patients than controls [0.64(0.31,1.24)AU vs 1.41(0.68,2.06)AU, p = 0.03, A] but not significantly different in COPD patients with a reduced FFMI compared to patients with a normal [0.73(0.34,1.61)AU vs 0.74(0.12,1.34)AU, p = 0.46, A]. There was a subset of COPD patients with relatively high protein levels of these mediators compared to the controls but these patients were not confined to the low FFMI group. MuRF-1 protein levels were not significantly different in quadriceps muscle from COPD patients compared to controls [0.56(0.45,1.30)AU vs 0.92(0.74,1.50)AU, p = 0.12, B], nor between COPD patients with and without a low FFMI [0.55(0.42,0.92)AU vs 0.59(0.54,1.62)AU, p = 0.35, B].](/cms/asset/9ccb3aff-4d82-43ae-b499-6f0706a94c2c/icop_a_781577_f0001_b.gif)
Figure 2. Representative images of western blots for atrogin-1 and MuRF-1 protein in COPD patients and controls. The atrogin-1 band and MuRF-1 bands were seen on separate blots just below the 50 kDA marker. The atrogin bands are denser in the control samples than the COPD samples, with considerable variability between patients, while the MuRF-1 bands show no consistent density difference between COPD samples and controls.
MuRF-1 protein in quadriceps was not significantly different between COPD patients and controls [0.56(0.45,1.30)AU vs 0.92(0.74,150)AU, p = 0.12, B] nor different between COPD patients with and without a low FFMI [0.55(0.42,0.92) vs 0.59(0.53, 1.62), p = 0.35, ]. These results were consistent with our findings from a larger group of COPD patients (n = 49) and controls (n = 23) overlapping with this current cohort in which we observed that atrogin-1 mRNA levels were significantly lower in patients compared to controls, while MuRF-1 mRNA levels were not significantly different between these groups, and neither atrogin-1 or MuRF-1 mRNA levels were different between patients with and without a low FFMI (see Tables E1 and E2 in supplement). In the COPD group there were no significant correlations between quadriceps MuRF-1 and atrogin-1 protein and FFMI, quadriceps type I or type II fiber CSA or quadriceps strength (whether measured as MVC or TwQ).
Discussion
The main finding of the present study is that MuRF-1 and atrogin-1 protein were not increased in the quadriceps of stable COPD patients with a reduced total muscle mass compared to patients with normal muscle mass. There were also no correlations between MuRF-1 and atrogin-1 protein and mRNA in the quadriceps with quadriceps fiber size. Similarly, MuRF-1 and atrogin-1 protein were not greater in the quadriceps of COPD patients compared to controls; in fact, controls had higher muscle atrogin-1 protein than patients. Therefore, our data are not supportive of a role of MuRF-1 and atrogin-1 in the maintenance of a chronic muscle-wasted state in stable patients with COPD.
Significance of the findings
Since our data point away from maintenance of a chronic wasted state being associated with increased muscle ubiquitin ligases, why then do patients sustain a low muscle mass? In chronic wasting in humans, impaired protein synthesis may be the driver of reduction in muscle mass with muscle protein breakdown rates actually becoming suppressed as an adaptation to limit muscle loss (Citation29). Our finding of reduced atrogin-1 protein expression in the muscle of COPD patients compared to controls is consistent with this concept. In cancer cachexia, which has similarities to COPD-related cachexia, the muscle loss is largely attributable to impaired muscle protein synthesis and not increased protein breakdown (Citation30, 31).
We acknowledge, however, that, despite our findings, strategies to block the action of atrogin-1 further may still be effective in treating muscle atrophy in COPD by shifting the balance further in favour of muscle protein synthesis. In addition, our data do not exclude the possibility that muscle atrophy results from acute catabolic episodes involving increases in MuRF-1 and atrogin-1, for example during an exacerbation, which cannot then be fully resolved during convalescence. Apart from exacerbation, an acute catabolic event could potentially be triggered by restarting smoking (Citation32) or stopping anti-inflammatory medication such as steroids, for example. In this case, blockade of these mediators during the catabolic episode may be effective in preventing development of a chronic wasted state.
Although our findings, at first glance, appear to contradict data from other studies (Citation12, 13), this is not the case. While the two prior studies are often cited, for example Kim et al. (Citation33) or Rabinovich and Vilario (Citation34), as evidence for a role for ubiquitin ligases in the quadriceps atrophy of COPD, they did not actually show any association between ubiquitin ligases and muscle atrophy in COPD. Doucet et al. did not find a difference in atrogin-1 protein levels in COPD patients and controls, only in mRNA levels, and did not measure MuRF-1 protein. As with our research, Doucet et al. did not find a difference in atrogin-1 or MuRF-1 mRNA levels between the 6 COPD patients with muscle wasting compared to 5 patients without wasting (Citation12).
Plant et al. found that MuRF-1 protein levels did not differ between muscle-wasted patients and healthy controls and did not measure atrogin-1 protein level (Citation13). The difference between Nedd4 protein levels in patients and controls could have been related to muscle wasting and/or COPD itself; the question could have been answered if Nedd4 had been compared in COPD patients with and without muscle wasting. However, our data, for the first time, expressly shows that muscle ubiquitin ligases are not increased in the specific context of stable muscle wasting in COPD.
Our data are also not inconsistent with animal models if timing of ubiquitin ligase measurement in relation to the incidence and nature of the insult is considered. In animal models of acute muscle wasting due to denervation or complete immobilisation, MuRF-1 and atrogin-1 mRNA levels peak 4 days after the insult then return to normal by day 14 (Citation4). Similarly, following immobilisation in humans, quadriceps MuRF-1 mRNA is 3-fold lower than baseline 20 days after the result, though it is increased 3-fold at day 10 (Citation35). In chronic wasting in humans, there is unlikely to be a single, sudden insult but a gradual accumulation of chronic factors (reduced physical activity, hypoxaemia, low-grade systemic inflammation), which means that ubiquitin ligases may not be upregulated in the same way as in animal models.
Lastly, the finding that atrogin-1 and MuRF-1 levels were not increased in patients with COPD compared to controls, nor specifically in patients with wasting, is consistent with our finding in patients from the same cohort that the P38 mitogen-activated kinase pathway was not activated in quadriceps (Citation10) and data from Mercken et al. showing that in clinically stable COPD, muscle NF-κB is not activated in resting conditions(Citation8).
Critique of the method
The study was designed to answer the question about whether MuRF-1 and atrogin-1 expression were different in COPD patients with and without muscle wasting, and investigated this in respect to both total muscle bulk and atrophy of individual muscle fiber types. Both MuRF-1 and atrogin-1 were quantified at a protein level, in contrast to previous studies. The present study includes data from COPD patients with a wider range of lung disease severity, which means our data is more generalizable to the COPD population than if we had selected only the very severe group. We did not specifically select GOLD III and IV patients, since it is known that the muscle weakness (Citation1) and wasting (Citation36) in COPD are not confined to those with the worst lung function We ensured, however, that severity of lung disease was matched between the patients with and without muscle wasting.
The data would have been strengthened had muscle protein breakdown and muscle protein synthesis rates also been measured, although these techniques are complex and would be difficult to accomplish in a sample of this size (Citation37).
One criticism of our study is that because we studied stable outpatients we were unlikely to find evidence of ubiquitin ligase activity, since this is a marker of active muscle atrophy. Although this is a pertinent criticism, it overlooks the following points. First, partially positive results had been reported prior to our study by Doucet et al. (Citation12) and Plant et al. (Citation13) in stable outpatients, supporting the validity of our study question. Second, while muscle wasting is almost certainly accelerated during exacerbation (see later), reduced muscle strength occurs in the absence of exacerbation and is unrelated to exacerbation frequency (Citation38).
In this cohort, exacerbation frequency was not significantly different between the patients with a low FFMI and patients with a normal FFMI (3.7 exacerbations per year versus 2.9 excaerbations per year respectively, p = 0.47). Last, we have recently demonstrated a relationship between muscle specific micro-RNA (miRNA) and muscle phenotype in COPD (Citation39); preliminary data confirm that muscle specific miRNA can also be detected in the blood of stable outpatients with COPD suggesting that active muscle atrophy is indeed occurring in apparently stable patients with COPD (Citation40).
Strength loss is a feature of acute exacerbation (Citation41), but we cannot comment on possible roles of MuRF-1 and atrogin-1 in loss of muscle mass at the time of an exacerbation (Citation38, Citation41) as we specifically excluded clinically unstable patients who had suffered a recent exacerbation. Our aim was to study clinically stable patients who were either maintaining or very slowly losing muscle mass. Data on any changes in patients’ weight and FFMI in the months preceding this study would have been helpful to define the exact nature of the patients (muscle mass maintaining or gradually decreasing) that this data relates to. Nevertheless, the cross-sectional study design we used, although also employed in the studies by Doucet et al. (Citation12) and Plant et al. (Citation13), would have been strengthened by a longitudinal arm.
In conclusion, we do not find any associations between MuRF-1 or atrogin-1 protein and global muscle atrophy, quadriceps muscle fiber size or quadriceps weakness in a group of patients with stable COPD. Our data do not support the concept that chronically elevated muscle levels of MuRF-1 or atrogin-1 are key drivers of muscle wasting in stable COPD.
Declaration of Interest Statement
S. A. Natanek (née Sathyapala) was funded by a Wellcome Trust Clinical Research Training Fellowship (079686) and was previously funded by GlaxoSmithKline. G.S. Marsh and J. Riddoch-Contreras were funded by an educational grant to Imperial College from GlaxoSmithKline. W. D.-C. Man is funded by a National Institute for Health Research Clinician Scientist Award (CS/7/2007). GlaxoSmithKline had no input into the data collection or manuscript preparation. The study was performed at the NIHR Respiratory Biomedical Research Unit of the Royal Brompton and Harefield NHS Trust and the National Heart and Lung Institute of Imperial College who also part-fund M. I. Polkey's salary.
SAN carried out the physiological testing, collection of muscle biopsies, the histological assays, the statistical analysis and drafted the manuscript, JRC helped with the laboratory experiments and writing of manuscript, GSM assisted SAN with collection of the muscle biopsies and physiological testing and interpretation of the data, NSH helped with troubleshooting for the physiological testing and drafting the manuscript, WD-CM and JM gave assistance with interpretation of the data and writing of the manuscript, and MIP and PRK supervised collection of the physiological data and laboratory analysis, respectively; both helped with data interpretation, conception of the idea and writing of the manuscript. All authors read and approved the final manuscript. Authors Kemp and Polkey contributed equally to this work.
Acknowledgements
We would like to thank Winston Banya, in the Statistics Department of the Royal Brompton Hospital, for his help with the statistical analysis, Derek Cramer, Mark Unstead and the lung function department at the Royal Brompton Hospital for performing the pulmonary function tests, Professor David Hansell and the CT department at the Royal Brompton Hospital for performing the thigh scans, and Kathleen Daenen at The University Hospital Maastricht for the cryosectioning.
Supplementary materials are available in the online version of this article.
References
- Seymour JM, Spruit MA, Hopkinson NS, Sathyapala SA, Man WD, Jackson A, Gosker HR, Schols AM, Moxham J, Polkey MI, The prevalence of quadriceps weakness in COPD and the relationship with disease severity. Eur Respir J 2010; 36:81–88.
- Marquis K, Debigare R, Lacasse Y, LeBlanc P, Jobin J, Carrier G, Maltais F. Midthigh muscle cross-sectional area is a better predictor of mortality than body mass index in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002; 166:809–813.
- Swallow EB, Reyes D, Hopkinson NS, Man WD, Porcher R, Cetti EJ, Moore AJ, Moxham J, Polkey MI. Quadriceps strength predicts mortality in patients with moderate to severe chronic obstructive pulmonary disease. Thorax 2007; 62:115–120.
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001; 294:1704–1708.
- Cai D, Frantz JD, Tawa NE, Jr., Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Ikkbeta/nf-kappab activation causes severe muscle wasting in mice. Cell 2004; 119:285–298.
- Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, Reid MB. Tnf-alpha acts via p38 mapk to stimulate expression of the ubiquitin ligase atrogin1/mafbx in skeletal muscle. FASEB J 2005; 19:362–370.
- Agusti A, Morla M, Sauleda J, Saus C, Busquets X. Nf-kappab activation and inos upregulation in skeletal muscle of patients with copd and low body weight. Thorax 2004; 59:483–487.
- Mercken EM, Hageman GJ, Langen RC, Wouters EF, Schols AM. Decreased exercise-induced expression of nuclear factor-kappab-regulated genes in muscle of patients with copd. Chest 2011; 139:337–346.
- Natanek SA LR, Gosker H R, Marsh GS, Slot IGMS, Hopkinson NS, Man WD-C, Moxham J, Kemp PR, Schols AMWJ, Polkey MI. Nf-kappa b (nf-κb) and activator protein-1 (ap-1) DNA binding in the quadriceps of copd patients. Thorax 2010; 65:A138–A139.
- Riddoch-Contreras J, George T, Natanek SA, Marsh GS, Hopkinson NS, Tal-Singer R, Kemp P, Polkey MI. P38 mitogen-activated protein kinase is not activated in the quadriceps of patients with stable chronic obstructive pulmonary disease. COPD 2012:142–150.
- Lemire BB, Debigare R, Dube A, Theriault ME, Cote CH, Maltais F. Mapk signalling in the quadriceps of patients with chronic obstructive pulmonary disease. J Appl Physiol 2012; 113(1):159–166.
- Doucet M, Russell AP, Leger B, Debigare R, Joanisse DR, Caron MA, LeBlanc P, Maltais F. Muscle atrophy and hypertrophy signaling in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007; 176:261–269.
- Plant PJ, Brooks D, Faughnan M, Bayley T, Bain J, Singer L, Correa J, Pearce D, Binnie M, Batt J. Cellular markers of muscle atrophy in chronic obstructive pulmonary disease (COPD). Am J Respir Cell Mol Biol 2010; 424:461–471.
- Schols AM, Soeters PB, Dingemans AM, Mostert R, Frantzen PJ, Wouters EF. Prevalence and characteristics of nutritional depletion in patients with stable copd eligible for pulmonary rehabilitation. Am Rev Respir Dis 1993; 147:1151–1156.
- Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Nhlbi/who global initiative for chronic obstructive lung disease (GOLD) workshop summary. Am J Respir Crit Care Med 2001; 163:1256–1276.
- Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, Crapo R, Enright P, van der Grinten CP, Gustafsson P, Standardisation of spirometry. Eur Respir J 2005; 26:319–338.
- Wanger J, Clausen JL, Coates A, Pedersen OF, Brusasco V, Burgos F, Casaburi R, Crapo R, Enright P, van der Grinten CP, Standardisation of the measurement of lung volumes. Eur Respir J 2005; 26:511–522.
- Macintyre N, Crapo RO, Viegi G, Johnson DC, van der Grinten CP, Brusasco V, Burgos F, Casaburi R, Coates A, Enright P, Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur Respir J 2005; 26:720–735.
- Pitta F, Troosters T, Spruit MA, Decramer M, Gosselink R. Activity monitoring for assessment of physical activities in daily life in patients with chronic obstructive pulmonary disease. Arch Phys Med Rehabil 2005; 86:1979–1985.
- Steiner MC, Barton RL, Singh SJ, Morgan MD. Bedside methods versus dual energy X-ray absorptiometry for body composition measurement in COPD. Eur Respir J 2002; 19:626–631.
- Edwards RH, Young A, Hosking GP, Jones DA. Human skeletal muscle function: Description of tests and normal values. Clin Sci Mol Med 1977; 52:283–290.
- Polkey MI, Kyroussis D, Hamnegard CH, Mills GH, Green M, Moxham J. Quadriceps strength and fatigue assessed by magnetic stimulation of the femoral nerve in man. Muscle Nerve 1996; 19:549–555.
- Ats statement: Guidelines for the six-minute walk test. Am J Respir Crit Care Med 2002; 166:111–117.
- Hopkinson NS, Man WD, Dayer MJ, Ross ET, Nickol AH, Hart N, Moxham J, Polkey MI. Acute effect of oral steroids on muscle function in chronic obstructive pulmonary disease. Eur Respir J 2004; 24:137–142.
- J B. Percutaneous needle biopsy of skeletal muscle in physiological and clinical research. Scand J Clin Lab Invest 1975; 35:609–616.
- Russell AP, Wadley G, Hesselink MK, Schaart G, Lo S, Leger B, Garnham A, Kornips E, Cameron-Smith D, Giacobino JP, Ucp3 protein expression is lower in type i, iia and iix muscle fiber types of endurance-trained compared to untrained subjects. Pflugers Arch 2003; 445:563–569.
- Verdijk LB, Koopman R, Schaart G, Meijer K, Savelberg HH, van Loon LJ. Satellite cell content is specifically reduced in type ii skeletal muscle fibers in the elderly. Am J Physiol Endocrinol Metab 2007; 292:E151–157.
- Natanek S.A, Gosker HR, Slot IGM, Marsh GS, Hopkinson NS, Man WD-C, Tal-Singer RM, Moxham J, Kemp PR, Schols AMWJ, Heterogeneity of quadriceps muscle phenotype in chronic obstructive pulmonary disease (copd); implications for stratified medicine? Muscle Nerve 2012; in press still PMID 23553751.
- Rennie MJ. Muscle protein turnover and the wasting due to injury and disease. Br Med Bull 1985; 41:257–264.
- Rennie MJ, Edwards RH, Emery PW, Halliday D, Lundholm K, Millward DJ. Depressed protein synthesis is the dominant characteristic of muscle wasting and cachexia. Clin Physiol 1983; 3:387–398.
- Rennie MJ, Harrison R. Effects of injury, disease, and malnutrition on protein metabolism in man. Unanswered questions. Lancet 1984; 1:323–325.
- Petersen AM, Magkos F, Atherton P, Selby A, Smith K, Rennie MJ, Pedersen BK, Mittendorfer B. Smoking impairs muscle protein synthesis and increases the expression of myostatin and mafbx in muscle. Am J Physiol Endocrinol Metab 2007; 293:E843–848.
- Kim HC, Mofarrahi M, Hussain SN. Skeletal muscle dysfunction in patients with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2008; 3:637–658.
- Rabinovich RA, Vilaro J. Structural and functional changes of peripheral muscles in chronic obstructive pulmonary disease patients. Curr Opin Pulm Med 2010; 16:123–133.
- de Boer MD, Selby A, Atherton P, Smith K, Seynnes OR, Maganaris CN, Maffulli N, Movin T, Narici MV, Rennie MJ. The temporal responses of protein synthesis, gene expression and cell signalling in human quadriceps muscle and patellar tendon to disuse. J Physiol 2007; 585:241–251.
- Shrikrishna D, Patel M, Tanner RJ, Seymour JM, Connolly BA, Puthucheary ZA, Walsh SL, Bloch SA, Sidhu PS, Hart N, Quadriceps wasting and physical inactivity in patients with copd. Eur Respir J 40(5):1115–1122.
- Rennie MJ, Smith K, Watt PW. Measurement of human tissue protein synthesis: An optimal approach. Am J Physiol 1994; 266:E298–307.
- Hopkinson NS, Tennant RC, Dayer MJ, Swallow EB, Hansel TT, Moxham J, Polkey MI. A prospective study of decline in fat free mass and skeletal muscle strength in chronic obstructive pulmonary disease. Respir Res 2007; 8:25.
- Lewis A, Riddoch-Contreras J, Natanek SA, Donaldson A, Man WD, Moxham J, Hopkinson NS, Polkey MI, Kemp PR. Downregulation of the serum response factor/mir-1 axis in the quadriceps of patients with COPD. Thorax 2012; 67:26-34.
- Donaldson AV, Natanek SA, Man WD-C, Kemp PR and Polkey MI. Increased skeletal muscle-specific microrna-1 in the blood of COPD patients. Thorax 2012; 65:A25.
- Spruit MA, Gosselink R, Troosters T, Kasran A, Gayan-Ramirez G, Bogaerts P, Bouillon R, Decramer M. Muscle force during an acute exacerbation in hospitalised patients with copd and its relationship with cxcl8 and igf-i. Thorax 2003; 58:752–756.