
Abstract
In chronic obstructive pulmonary disease (COPD), two major pathological changes that occur are the loss of alveolar structure and airspace enlargement. Type II alveolar epithelial cells (AECII) play a vital role in maintaining alveolar homeostasis and lung tissue repair. Sirtuin 1 (SIRT1), a NAD+-dependent histone deacetylase, regulates many pathophysiological processes including inflammation, apoptosis, cellular senescence and stress resistance. The main aim of this study was to investigate whether SRT1720, a pharmacological SIRT1 activator, could protect against AECII apoptosis in rats with emphysema caused by cigarette smoke exposure and intratracheal lipopolysaccharide instillation in vivo. During the induction of emphysema in rats, administration of SRT1720 improved lung function including airway resistance and pulmonary dynamic compliance. SRT1720 treatment up-regulated the levels of surfactant protein (SP)A, SPC, SIRT1 and forkhead box O 3, increased SIRT1 activity, down-regulated the level of p53 and inhibited AECII apoptosis. Lung injury caused by emphysema was alleviated after SRT1720 treatment. SRT1720 could protect against AECII apoptosis in rats with emphysema and thus could be used in COPD treatment.
Introduction
In COPD, the major mechanisms underlying chronic airflow limitation are the perpetual loss of alveolar structure, which causes airspace enlargement, and the destruction of the lung parenchyma and the loss of elastic recoil (Citation1, 2). In the progression of COPD, a key role is played by the apoptosis of lung epithelial cells, which results in the destruction of the alveolar structure and in emphysema (Citation3, 4). In mammals, the alveolar epithelium is composed of type I alveolar epithelial cells (AECI) and type II alveolar epithelial cells (AECII). AECII are the multifunctional secretory cells that are characterized by the presence of morphological lamellar bodies containing pulmonary surfactant proteins (SPs) and lipids. AECII play vital roles in synthesizing and secreting lung surfactants, which include SPA, SPB, SPC, and SPD, reducing the surface tension of the alveoli, maintaining normal alveolar homeostasis and gas exchange, and improving lung tissue repair (Citation5, 6).
Sirtuin 1 (SIRT1) is a member of the sirtuin proteins family, which is a NAD+-dependent class III histone deacetylase (Citation7, 8). SIRT1, a nuclear protein in mammals, deacetylates histone and transcription factors (regulating signal pathways of inflammation, cellular senescence, stress resistance, apoptosis and proliferation), such as forkhead box O3 (FoxO3), p53 and nuclear factor-κB (Citation9–11). As a selective pharmacological activator of SIRT1, SRT1720 exhibits effective pathophysiological functions.
On the one hand, the therapeutic effects of SRT1720 have been investigated in several diseases such as inflammation in asthma (Citation12), diabetes (Citation13, 14), cardiovascular disease (Citation15), kidney injury (Citation16) and COPD (Citation17, 18). Yao et al. (Citation19) reported that SIRT1 activator SRT1720 protects against emphysema through reduction of cellular senescence in mice. On the other hand, SRT1720 improves the resistance of acute kidney injury and promotes longevity of obese mice via the suppression of apoptosis (Citation16, Citation20).
However, it is still unknown whether SRT1720 could alleviate lung injury through suppression of AECII apoptosis. In this study, we investigated whether SRT1720 inhibited AECII apoptosis in rats with emphysema and alleviated lung injury in vivo.
Materials and Methods
Animal model
We purchased 45 male Sprague–Dawley (SD) rats (body weight: 200–220 g) from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China). We maintained the rats at 21ºC–25ºC and 40%–60% relative humidity in a 12-hour light/dark cycle and provided them with water and food ad libitum in the Laboratory Animal Center of Zhejiang Chinese Medical University (Hangzhou, China). The rats were randomly sorted into 3 groups (15 rats/group): control (group I), emphysema (group II) and emphysema+SRT1720 (group III). All animal protocols were approved by the Institutional Animal Care and Use Committee of Zhejiang Chinese Medical University.
A custom-designed cigarette smoking chamber (Citation21) and lipopolysaccharide (LPS; Sigma, St. Louis, MO, USA) stimuli were used for generating the rat model of emphysema (Citation22). In this study, we used commercially available cigarettes (XiongShi; China Tobacco Zhejiang Industrial. Co., Ltd., China) that contained 0.7 mg of nicotine and 8 mg of tar per cigarette. Briefly, cigarette smoke exposure and intratracheal LPS instillation were performed as follows: rats in groups II and III were placed in the cigarette smoking chamber (60 cm × 50 cm × 40 cm), and after the rats had settled, the smoke of 5 cigarettes was successively delivered in 12 minutes.
After an interval of 10 minutes, the smoke of 5 new cigarettes was delivered into the chamber. The rats were exposed to 20 cigarettes over 90 minutes once a day for each smoke exposure and for 7 days per week for 12 weeks. During the exposure, the concentration of carbon monoxide was maintained almost constant: after 30, 60, and 90 minutes’ exposures, the concentrations were 402 ± 19, 399 ± 12, and 408 ± 14 ppm, respectively, as measured using a carbon monoxide detector (CTB-999; Industrial Scientific Co., Ltd., Shanghai, China).
Meanwhile, the smoke concentration was set at ∼300 mg/m3 total particulate matter (TPM) in the cigarette smoking chamber, monitored by a real time aerosol monitor (MicroDust Pro, Casella CEL, Bedford, UK) (Citation19). On the last day of the 4th and 8th weeks, each rat in groups II and III was temporarily anesthetized with 5.0% isoflurane, after which the rats were intratracheally instilled with 200 μL of 1 μg/μL LPS in sterile phosphate-buffered solution (PBS). The rats in group I received clean throughout the experimental period and were intratracheally instilled 200 μL sterile normal saline at the same time.
Administration of SRT1720
To investigate the therapeutic effects on rats with emphysema, SRT1720 was administered as described previously (Citation19). After the second intratracheal LPS instillation, each rat in group III was treated with SRT1720 (100 mg/kg body weight in 2% HPMC and 0.2% DOSS, >95% pure by C-13 nuclear magnetic resonance and liquid chromatography-mass spectrometry, synthesized by Life Chemicals, Inc., Burlington, Canada) through oral gavage which was repeated daily, 1 hour prior to smoke exposure for 4 weeks.
Lung function determination
Ten rats in each group were randomly chosen for lung function determination including airway resistance (AR) and pulmonary dynamic compliance (Cdyn), 24 hours after the last smoke exposure. Briefly, the rats were continuously anesthetized with 5.0% isoflurane and tracheostomized. The Y type endotracheal trachea was cannulated and the esophageal cannula was intubated to the middle of the esophagus. Then, the endotracheal trachea was connected to the micro-pressure transducer and the respiratory flow transducer, and the esophageal cannula was connected to the pressure transducer (Shanghai Alcott Biotech Co., Ltd., Shanghai, China). After stabilized, the signal waves were collected continuously for 10 minutes.
The parameters of AR and Cdyn, which analyzed 100 respiratory cycles were measured using a MPA lung function system (Shanghai Alcott Biotech Co., Ltd.). And then, rats were sacrificed after the lung function determination by intraperitoneal injection of 10.0% chloral hydrate. The left lungs were removed and fixed in 4% paraformaldehyde for histological examination and immunohistochemical staining. The right lungs were stored at −80°C for use in PCR, western blotting and SIRT1 activity analyses.
Lung histology
The left lungs were perfused intratracheally with 4% paraformaldehyde at a constant pressure of 25 cm H2O for 1 hour, and then immersed in paraformaldehyde for 24 hours to fix them completely. After fixation, the left-lung blocks were dehydrated and embedded in paraffin and cut into 4-μm-thick sections using a rotary microtome (HM315R, Microm, Germany). Three discontinuous paraffin-embedded sections of each lung sample were stained with hematoxylin and eosin (H&E) in order to assess the morphological changes in the lungs.
We examined 5 fields of view in each of the 3 sections from each lung sample by using a light microscope (Olympus, Tokyo, Japan), and we avoided selecting fields containing bronchi and large blood vessels. We obtained the mean linear intercept (MLI), which indicates the average distance between opposing walls of a single alveolus and is a measure of pulmonary airspace enlargement; moreover, we obtained the mean alveolar airspace (MAA), which is also a measure of pulmonary airspace enlargement (Citation21, Citation23).
Quantitative real-time PCR
Total RNA was extracted from each right-lung sample by using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocols. The final RNA purity and concentrations were determined using a spectrophotometer. Next, cDNA was synthesized from the total RNA by using the PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa, Otsu, Janpa). Quantitative real-time PCR analysis was carried out using previously described parameters (Citation24).
The following primers were used (Invitrogen, Shanghai, China): SPA, 5’-TCGGTGTCCCAGGATTTAG-3’, 5’-CAGGGTGGCTGCTGTTAGT-3’; SPC, 5’-CAGACACCATCGCTACCTT-3’, 5’-TAGCCAAAGCCTCA AGACT-3’; GAPDH, 5’-GTTCAACGGCACAGTCA AG-3’, 5’-GCCAGTAGACTCCACGACAT-3’. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a reference gene. All analyses were performed in triplicate. The relative expression of SPA and SPC mRNA was calculated using the 2−∆∆Ct method.
Immunohistochemistry
We performed immunohistochemical staining in order to examine the expression of SPA and SPC proteins in formalin-fixed, paraffin-embedded sections of each left-lung sample. Sections were deparaffinized and rehydrated using a graded series of ethanol, and this was followed by a high-pressure antigen-retrieval step. Endogenous peroxidase activity was blocked by incubating sections with 3% hydrogen peroxide for 15 minutes. The sections were next blocked with 10% goat serum (ZSGB-BIO, Beijing, China) for 1 hour, incubated (overnight, 4°C) with rabbit anti-rat SPA or SPC primary antibodies (1:500; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), and then stained with an anti-rabbit IgG secondary antibody (SP-9000 kit; ZSGB-BIO) and 3,3’-diaminobenzidine (DAB; ZSGB-BIO). Hematoxylin was used for counterstaining. To calculate the percentages of SPA and SPC-positive cells, we used a light microscope and counted the SPA and SPC-positive cells present among a total of 400 cells in 10 different lung samples, and we used 10 randomly selected alveolus fields in the case of each lung sample.
TUNEL/SPC immunofluorescence staining
AECII apoptosis in each group was detected using terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL; Beyotime)/SPC double-staining as described (Citation25). Paraffin-embedded left-lung sections were deparaffinized and rehydrated and then subjected to the high-pressure antigen-retrieval step and proteinase K (20 μg/mL) treatment for 20 minutes. The sections were next incubated with the rabbit anti-rat SPC primary antibody (1:300; Santa Cruz) overnight at 4°C and then with the FITC-conjugated anti-rabbit IgG secondary antibody (1:500; Santa Cruz) for 1 hour at 37°C, and this was followed by incubation with 50 μL of the TUNEL reaction mixture at 37˚C for 1 hour in a humidified chamber.
Finally, the sections were counterstained with 4’,6-diamino-2-phenylindole (DAPI; Sigma) and examined using an IX71 fluorescence microscope (Olympus). We randomly selected 100 images from 10 different lung samples, and then chose 10 discontinuous areas of each lung sample for the analysis. The percentage of AECII apoptosis was determined by dividing the number of apoptotic AECII, which were positive for both TUNEL and SPC, by the total number of DAPI-positive cells.
Western blot and SIRT1 activity assay
Proteins were extracted from each right-lung sample by homogenization in ice-cold lysis buffer (50 mmol/L Tris, pH 7.4; 150 mmol/L NaCl, 0.1% sodium dodecyl sulfate, 1 mmol/L EDTA, 1% sodium deoxycholate and 1% Triton X-100) containing a protease inhibitor cocktail (1 mmol/L phenylmethylsulfonyl fluoride, 1 mg/L leupeptin and 1 mg/L aprotinin). Then homogenates were centrifuged for 15 minutes at 12,000 rpm and the supernatants were collected. The protein concentration in samples was determined using a micro BCA protein assay kit (Pierce, Rockford, IL, USA), according to the manufacturer's protocols.
The equal amounts of protein (20 μg) were heated at 100°C for 5 minutes and then separated by electrophoresing them on 8% sodium dodecyl sulfate-polyacrylamide gels. Then separated proteins were electrotransferred to nitrocellulose membranes, blocked with Tris-buffered saline with Tween-20 (TBS-T) solution containing 5% bovine serum albumin (BSA) for 2 hours at room temperature. Membranes were incubated with primary antibodies (1:1000 dilution in TBS-T) of SIRT1, FoxO3, p53 (Cell Signaling Technology, Beverly, MA, USA) and β-actin (Abcam, Cambridge, MA, USA) overnight at 4°C on an orbital shaker.
After washed 3 times for 10 minutes each in TBS-T, the membranes were then incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG (H+L) (Pierce) for 1 hour at room temperature on an orbital shaker. After washing 5 times with TBS-T, bands were detected using an enhanced chemiluminescence solution (Amersham Pharmacia Biotech, Buckinghamshire, UK), visualized by X-ray film exposure and analyzed using a UVP-GDS8000 gel analysis system (Ultra-Violet Products Ltd., United Kingdom). Protein expression levels were analyzed by performing densitometry and the values were normalized relative to those for β-actin, which was used as the loading control.
SIRT1 activity was analyzed using a deacetylase colorimetric activity assay kit (Enzo Life Sciences, Farmingdale, NY, USA) according to the manufacturer's protocols. Briefly, SIRT1 immunoprecipitation from right lung sample extracts was performed as described (Citation26). The Color de Lys substrate and NAD+ was added to the SIRT1-conjugated beads and incubated at 37°C for 1 hour. Then, the substrate-SIRT1 mixture was added to a 96-well plate followed by addition of the Color de Lys developer for 15 minutes at 37°C. Finally, the plate was read by a microtiter-plate reader (M200, Tecan Group Ltd., Switzerland) at 405 nm.
Statistical analyses
Statistical analyses were performed using a SPSS 17.0 software. Data are expressed as means ± SD. The significance of difference between groups was determined by means of one-way analysis of variance (ANOVA) and independent-sample t-tests. Multiple comparisons in ANOVA were performed using the Student-Newman-Keuls test. p < 0.05 was considered statistically significant.
Results
Lung function changes
AR was increased markedly and Cdyn was decreased in emphysema group (group II) after cigarette smoke exposure and intratracheal LPS instillation, which was consistent with emphysema characteristics of lung function compared to control group (group I) (p < 0.01). However, after administration of SRT1720, AR and Cdyn was markedly improved in group III as compared with group II (p < 0.01) (Table ).
Table 1. Alteration of airway resistance (AR) and pulmonary dynamic compliance (Cdyn) in rats from each group
Histological changes
After the last cigarette smoke exposure, histological changes were analyzed by H&E staining. In group II, enlargement of airspace was observed and the amount of alveoli was decreased in the lung tissues (Figure ). Compared with control group (Figure ), many alveoli were merged and some bullae were formed in group II, which was consistent with the pathological characteristics of emphysema. However, as shown in Figure , the characteristics of emphysema were reduced after rats treated with SRT1720. Quantitative analyses of lung histomorphology revealed that the MLI and MAA of group II was higher than those of the control group (group I) (p < 0.05). Administration of SRT1720 in group III, MLI and MAA was decreased obviously compared to group II (p < 0.05) (Table ).
Figure 1. Histological examination of lung tissues. Airspace were enlarged obviously and the amount of alveoli was decreased after cigarette smoke exposure and intratracheal LPS instillation in emphysema group compared to control group. The double arrows indicate merged alveoli, and n = 10 for each group. The characteristics of emphysema were reduced after rats treated with SRT1720 in emphysema+SRT1720 group. Magnification, ×200.

Table 2. Quantitative analyses of lung histomorphology
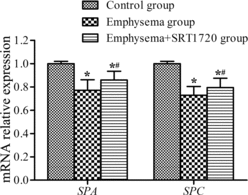
mRNA expression of SPA and SPC
High levels of SPA and SPC mRNAs were expressed in samples from group I (control group), but these levels were markedly lower in the emphysema group (group II) (p < 0.05). After administration of SRT1720 (group III), the expression levels of SPA and SPC mRNAs were significantly higher than those measured in the case of group II (p < 0.05) (Figure ).
Figure 2. Surfactant protein (SP)A and SPC mRNA expression in lung tissues. SPA and SPC mRNA expression in lung tissues of each group was analyzed by quantitative Real-Time polymerase chain reaction (PCR). All analyses were performed in triplicate. The relative expression of SPA and SPC mRNA was calculated with the 2−∆∆Ct method. Values are presented as mean ± SD (n = 10). *p < 0.05 vs. control group; #p < 0.05 vs. emphysema group.
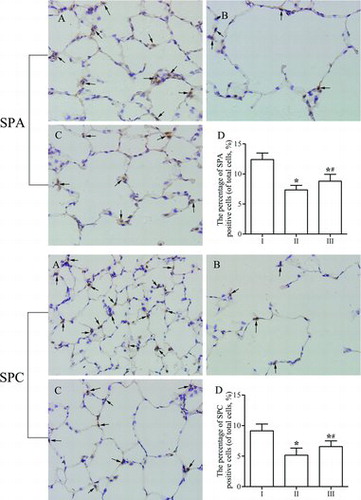
Immunohistochemical analysis of SPA and SPC expression
After immunohistochemical staining, SPA- and SPC-positive cells exhibited a yellowish-brown color in the cytoplasm, and these cells were mainly located in the alveolar corner of AECII (Figures –C). As shown in Figure , the expression of SPA and SPC was weaker in group II than in group I (Figure ). However, after SRT1720 administration (group III), the expression of SPA and SPC increased significantly compared with that in the emphysema group (group II). Similarly, the percentages of SPA- and SPC-positive cells were markedly lower in group II than in group I (p < 0.05; Figure ). However, the percentages of SPA- and SPC-positive cells were significantly higher in group III than in group II (p < 0.05). The results indicate that SRT1720 could protect against reduction of SPA and SPC expression in the development of emphysema, which was consistent with the expression of SPA and SPC mRNA in lung tissues.
Figure 3. Immunohistochemical analysis of the expression of surfactant protein (SP)A and SPC in lung parenchyma. (A) Control group. SPA and SPC staining was strongly positive with dark brown in cytoplasm. (B) Emphysema group. SPA and SPC expression was more faint than control group. (C) Emphysema+SRT1720 group. SRT1720 administration increased the expression of SPA and SPC as compared with emphysema group. The arrows indicate the SPA or SPC positive cells. Magnification, ×400. (D) The percentage of SPA or SPC positive cells in each group. Values are presented as mean ± SD (n = 10). *p < 0.05 vs. control group; #p < 0.05 vs. emphysema group.
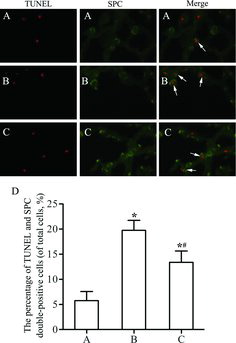
Apoptosis of type II alveolar epithelial cells
AECII apoptosis was detected using TUNEL and SPC double-immunofluorescence staining. Apoptotic AECII exhibited red fluorescence in the cell nuclei (TUNEL-positive) and green fluorescence in the cytoplasm (SPC-positive) (Figure –C). The percentages of AECII apoptosis in groups II and III were significantly higher than that in group I (p < 0.05; Figure ). After SRT1720 treatment (group III), the percentage of AECII apoptosis was markedly lower than that in group II (p < 0.05), which was consistent with the expression levels of SPA and SPC. These results indicated that AECII apoptosis decreased in rats after SRT1720 treatment.
Figure 4. Apoptosis of type II alveolar epithelial cells (AECII) in lung tissues. (A) Control group. A few TUNEL (red) and SPC (green) double-positive cells (apoptotic AECII) were found infrequently. (B) Emphysema group. More apoptotic AECII were observed after cigarette smoke exposure and intratracheal LPS instillation. (C) Emphysema+SRT1720 group. The number of apoptotic AECII was decreased after SRT1720 administration compared to emphysema group. The arrows indicate the apoptotic AECII. Magnification, ×400. (D) The number of apoptotic AECII in each group. Values are presented as mean ± SD (n = 10). *p < 0.05 vs. control group; #p < 0.05 vs. emphysema group.
Protein expression and SIRT1 activity
To further investigate the mechanisms of suppression AECII apoptosis after SRT1720 treatment, Western blot analysis was used to analyze the expression levels of SIRT1, FoxO3 and p53. The expression of SIRT1 and FoxO3 was down-regulated, but p53 was up-regulated in emphysema group compared to control group (p < 0.05). However, SRT1720 treatment (group III) increased the expression levels of SIRT1 and FoxO3, down-regulated p53 expression compared to group II (p < 0.05) (Figure –B).
Figure 5. Western blot analysis of the expression of SIRT1, FoxO3 and p53. (A-B) Expression of SIRT1, FoxO3 and p53 in lung tissues was analyzed by western blot analysis. β-actin was provided as the loading control. The expression of protein was analyzed by densitometry and normalized with β-actin. (C) SIRT1 activity was assayed in lung tissues. All analyses were performed in triplicate. Values are presented as mean ± SD (n = 10). *p < 0.05 vs. control group; #p < 0.05 vs. emphysema group.
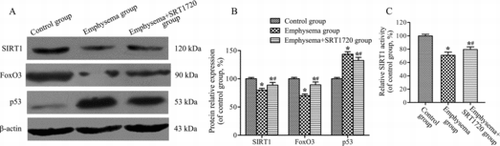
Furthermore, the SIRT1 activity was assayed in lung tissues to determine whether cigarette smoke exposure and intratracheal LPS instillation and SRT1720 treatment changed SIRT1 deacetylase activity. Accompanying the decrease in SIRT1 protein, there was a significant reduction of SIRT1 activity in group II in response to cigarette smoke exposure and intratracheal LPS instillation compared to group I (p < 0.05). However, administration of SRT1720 increased SIRT1 activity in group III contrarily (p < 0.05) (Figure ).
Discussion
COPD, which is considered to become the third leading cause of death worldwide by 2020, is currently recognized as a major global public health challenge. Cigarette smoking is the primary risk factor in the development of COPD (Citation27). LPS, which is a structural component of the outer membrane of gram-negative bacteria, plays key roles in injuring the epithelium and activating inflammatory cells to secrete inflammatory factors and proteases, which contribute to the pathogenesis of chronic bronchitis and emphysema (Citation28). In this study, emphysema was induced in rats by exposure to cigarette smoke and LPS.
As progenitors of AECI, AECII play vital roles in synthesizing and secreting lung surfactants, which include SPA, SPB, SPC, and SPD, reducing the surface tension of the alveoli, maintaining normal alveolar homeostasis and gas exchange, and improving lung tissue repair (Citation5, Citation29). The apoptosis of alveolar epithelial cells in lungs is an important pathogenesis of COPD and emphysema (Citation25, Citation30, Citation31). Lung function loss and airflow limitation are relevant to lung injury in rats with emphysema, which reflects the level of airway obstruction caused by the loss of alveolar structure, swollen bronchial walls and mucus hypersecretion (Citation21).
The lung function of rats exposed to cigarette smoke and intratracheally instilled with LPS was evaluated. The markers for airflow obstruction and lung elasticity, AR and Cdyn, changed significantly after cigarette smoke exposure and intratracheal LPS instillation, which are indicative of the extent of emphysema in the lung. After SRT1720 administration, the lung function and morphological changes of the lung tissues were examined. The results revealed that AR and Cdyn, MLI and MAA were obviously improved compared to emphysema group, which was consistent with the findings by Yao et al. (Citation19).
Thus, we hypothesized that SRT1720 administration might improve the lung function and attenuate lung injury in rats with emphysema, which may be due to suppress apoptosis of AECII. Cigarette smoke exposure induced apoptosis of alveolar cells and cellular senescence and caused air space enlargement (Citation25, Citation32). To locate and count the apoptotic AECII in the lung tissues, we performed TUNEL/SPC immunofluorescence staining. The results revealed that the percentage of apoptotic AECII was decreased after SRT1720 treatment compared with emphysema group.
Previous studies have shown that the level of SIRT1 is decreased in lung tissues of patients with COPD or murine exposed to cigarette smoke (Citation19, Citation26, Citation33), which is also associated with cellular senescence in the development of emphysema (Citation19). SIRT1 is recognized as a vital survival factor and improves the resistance in mice of acute kidney injury, regulating the suppression of cell apoptosis in the kidney (Citation16).
However, the molecular mechanisms of the SIRT1 protective effects on lung injury remain elusive. The imbalance of tissue inhibitor of matrix metalloproteinases (TIMPs)/matrix metalloproteinases (MMPs) (Citation34–36) and oxidative stress (Citation37, 38) in the lungs play a vital role in the development of COPD and emphysema. As Yao et al reported, SIRT1 protects against COPD and emphysema by redressing the imbalance of TIMP-1/MMP-9 including TIMP-1 deacetylation (Citation39), and also protects against cigarette smoke-induced lung oxidative stress mediated by FoxO3 (Citation40). The role of SIRT1 in tumorigenesis is controversial, although Zhang et al. (Citation41) and Chen et al. (Citation42) reported that SIRT1 expression may be a marker of the clinical features and prognosis in advanced non-small cell lung cancer, and plays a stimulative role in tumorigenesis of lung adenocarcinoma.
However, the role of SIRT1 in cigarette smoke and LPS-induced AECII apoptosis is still unclear. It is known that SIRT1 activity in lungs of rats is down-regulated by cigarette smoke exposure. Caito et al. (Citation26) confirmed that SIRT1 activity in lung epithelial cells was reduced by carbonyl and oxidative stress in response to cigarette smoke extract. p53, a tumor suppressor protein, possesses potential to protect from DNA damage by motivating DNA repair or inducing cell apoptosis after DNA damage exceeded a certain threshold (Citation43, 44).
SIRT1 has biochemical abilities to deacetylate lysine residues of p53 or regulate the promoter of p53 gene, which are due to inactivated p53 activity and reduce cellular senescence (Citation45). As an important member of forkhead box O subfamily, FoxO3 is a non-histone substrate of SIRT1. Combining SIRT1 and phosphorylated FoxO3 could form the protein complexes which regulate cellular senescence and apoptosis (Citation46).
In this study, the expression of SIRT1 protein and activity and FoxO3 was decreased, and p53 level was up-regulated, after rats were induced to be exposed cigarette smoke and intratracheally instilled LPS. However, SRT1720 treatment increased the expression of SIRT1 protein and activity and FoxO3, but down-regulated p53 expression.
Conclusion
SRT1720 treatment increased SIRT1 level and activity, which is associated with up-regulated FoxO3 expression, down-regulated p53 expression and inhibited apoptosis of AECII, suggesting that SRT1720 might regulate beneficial effects on emphysema by suppressing apoptosis. However, the further molecular mechanisms of the SIRT1 and SIRT1 activation protective effects on lung injury of emphysema by suppressing apoptosis of AECII remain elusive, which is imperative to explore in the near future.
Declaration of Interest Statement
This study was supported by grants from the National Natural Science Foundation of China (No. 81000016 and 81470241). The authors declare that there is no conflict of interest. The authors are responsible for the content and writing of this paper.
References
- Mizutani N, Fuchikami J, Takahashi M, Nabe T, Yoshino S, Kohno S. Pulmonary emphysema induced by cigarette smoke solution and lipopolysaccharide in guinea pigs. Biol Pharm Bull 2009; 32(9):1559–1564.
- Burgel PR. Pathogenesis of chronic obstructive pulmonary disease. Presse Med 2009; 38(3):406–412.
- Siganaki M, Koutsopoulos AV, Neofytou E, Vlachaki E, Psarrou M, Soulitzis N, Pentilas N, Schiza S, Siafakas NM, Tzortzaki EG. Deregulation of apoptosis mediators’ p53 and bcl2 in lung tissue of COPD patients. Respir Res 2010; 11:46.
- Zhao CZ, Fang XC, Wang D, Tang FD, Wang XD. Involvement of type II pneumocytes in the pathogenesis of chronic obstructive pulmonary disease. Respir Med 2010; 104(10):1391–1395.
- Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu Rev Med 2010; 61:105–119.
- Hoffman AM, Ingenito EP. Alveolar epithelial stem and progenitor cells: emerging evidence for their role in lung regeneration. Curr Med Chem 2012; 19(35):6003–6008.
- Milne JC, Denu JM. The Sirtuin family: therapeutic targets to treat diseases of aging. Curr Opin Chem Biol 2008; 12(1):11–17.
- Bordo D. Structure and evolution of human sirtuins. Curr Drug Targets 2013; 14(6):662–665.
- Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins–novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov 2008; 7(10):841–853.
- Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature 2009; 460(7255):587–591.
- Satoh A, Stein L, Imai S. The role of mammalian sirtuins in the regulation of metabolism, aging, and longevity. Handbk Exp Pharmacol 2011; 206:125–162.
- Ichikawa T, Hayashi R, Suzuki K, Imanishi S, Kambara K, Okazawa S, Inomata M, Yamada T, Yamazaki Y, Koshimizu Y, Miwa T, Matsui S, Usui I, Urakaze M, Matsuya Y, Sasahara M, Tobe K. Sirtuin 1 activator SRT1720 suppresses inflammation in an ovalbumin-induced mouse model of asthma. Respirology 2013; 18(2):332–339.
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007; 450(7170):712–716.
- Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab 2008; 8(5):347–358.
- Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res 2007; 100(10):1512–1521.
- Fan H, Yang HC, You L, Wang YY, He WJ, Hao CM. The histone deacetylase, SIRT1, contributes to the resistance of young mice to ischemia/reperfusion-induced acute kidney injury. Kidney Int 2013; 83(3):404–413.
- Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2008; 177(8):861–870.
- Rahman I, Kinnula VL, Gorbunova V, Yao H. SIRT1 as a therapeutic target in inflammaging of the pulmonary disease. Prev Med 2012; 54 Suppl:S20–S28.
- Yao H, Chung S, Hwang JW, Rajendrasozhan S, Sundar IK, Dean DA, McBurney MW, Guarente L, Gu W, Rönty M, Kinnula VL, Rahman I. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J Clin Invest 2012; 122(6):2032–2045.
- Minor RK, Baur JA, Gomes AP, Ward TM, Csiszar A, Mercken EM, Abdelmohsen K, Shin YK, Canto C, Scheibye-Knudsen M, Krawczyk M, Irusta PM, Martín-Montalvo A, Hubbard BP, Zhang Y, Lehrmann E, White AA, Price NL, Swindell WR, Pearson KJ, Becker KG, Bohr VA, Gorospe M, Egan JM, Talan MI, Auwerx J, Westphal CH, Ellis JL, Ungvari Z, Vlasuk GP, Elliott PJ, Sinclair DA, de Cabo R. SRT1720 improves survival and healthspan of obese mice. Sci Rep 2011; 1:70.
- Zheng H, Liu Y, Huang T, Fang Z, Li G, He S. Development and characterization of a rat model of chronic obstructive pulmonary disease (COPD) induced by sidestream cigarette smoke. Toxicol Lett 2009; 189(3):225–234.
- Jiang H, Zhu Y, Xu H, Sun Y, Li Q. Activation of hypoxia-inducible factor-1alpha via nuclear factor-kappa B in rats with chronic obstructive pulmonary disease. Acta Biochim Biophys Sin (Shanghai) 2010; 42(7):483–488.
- Agarwal AR, Yin F, Cadenas E. Short-term cigarette smoke exposure leads to metabolic alterations in lung alveolar cells. Am J Respir Cell Mol Biol 2014; 51(2):284–293.
- Li Y, Xu W, Yan J, Xia Y, Gu C, Ma Y, Tao H. Differentiation of human amniotic fluid-derived mesenchymal stem cells into type II alveolar epithelial cells in vitro. Int J Mol Med 2014; 33(6):1507–1513.
- Farkas L, Farkas D, Warburton D, Gauldie J, Shi W, Stampfli MR, Voelkel NF, Kolb M. Cigarette smoke exposure aggravates air space enlargement and alveolar cell apoptosis in Smad3 knockout mice. Am J Physiol Lung Cell Mol Physiol 2011; 301(4):L391–401.
- Caito S, Rajendrasozhan S, Cook S, Chung S, Yao H, Friedman AE, Brookes PS, Rahman I. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J 2010; 24(9):3145–3159.
- Macnee W. Pathogenesis of chronic obstructive pulmonary disease. Clin Chest Med 2007; 28(3):479–513.
- Martin TR. Recognition of bacterial endotoxin in the lungs. Am J Respir Cell Mol Biol 2000; 23(2):128–132.
- Fujino N, Kubo H, Suzuki T, Ota C, Hegab AE, He M, Suzuki S, Suzuki T, Yamada M, Kondo T, Kato H, Yamaya M. Isolation of alveolar epithelial type II progenitor cells from adult human lungs. Lab Invest 2011; 91(3):363–378.
- Aoshiba K, Yokohori N, Nagai A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am J Respir Cell Mol Biol 2003; 28(5):555–562.
- Mimae T, Hagiyama M, Inoue T, Yoneshige A, Kato T, Okada M, Murakami Y, Ito A. Increased ectodomain shedding of lung epithelial cell adhesion molecule 1 as a cause of increased alveolar cell apoptosis in emphysema. Thorax 2014; 69(3):223–231.
- Hara H, Araya J, Takasaka N, Fujii S, Kojima J, Yumino Y, Shimizu K, Ishikawa T, Numata T, Kawaishi M, Saito K, Hirano J, Odaka M, Morikawa T, Hano H, Nakayama K, Kuwano K. Involvement of creatine kinase B in cigarette smoke-induced bronchial epithelial cell senescence. Am J Respir Cell Mol Biol 2012; 46(3):306–312.
- Nakamaru Y, Vuppusetty C, Wada H, Milne JC, Ito M, Rossios C, Elliot M, Hogg J, Kharitonov S, Goto H, Bemis JE, Elliott P, Barnes PJ, Ito K. A protein deacetylase SIRT1 is a negative regulator of metalloproteinase-9. FASEB J 2009; 23(9):2810–2819.
- Elkington PT, Friedland JS. Matrix metalloproteinases in destructive pulmonary pathology. Thorax 2006; 61(3):259–266.
- Boschetto P, Quintavalle S, Zeni E, Leprotti S, Potena A, Ballerin L, Papi A, Palladini G, Luisetti M, Annovazzi L, Iadarola P, De Rosa E, Fabbri LM, Mapp CE. Association between markers of emphysema and more severe chronic obstructive pulmonary disease. Thorax 2006; 61(12):1037–1042.
- Ilumets H, Rytilä P, Demedts I, Brusselle GG, Sovijärvi A, Myllärniemi M, Sorsa T, Kinnula VL. Matrix metalloproteinases -8, -9 and -12 in smokers and patients with stage 0 COPD. Int J Chron Obstruct Pulmon Dis 2007; 2(3):369–379.
- Macnee W, Rahman I. Oxidants and antioxidants as therapeutic targets in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999; 160(5 Pt 2):S58–S65.
- Yao H, Rahman I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol Appl Pharmacol 2011; 254(2):72–85.
- Yao H, Hwang JW, Sundar IK, Friedman AE, McBurney MW, Guarente L, Gu W, Kinnula VL, Rahman I. SIRT1 redresses the imbalance of tissue inhibitor of matrix metalloproteinase-1 and matrix metalloproteinase-9 in the development of mouse emphysema and human COPD. Am J Physiol Lung Cell Mol Physiol 2013; 305(9):L615–L624.
- Yao H, Sundar IK, Ahmad T, Lerner C, Gerloff J, Friedman AE, Phipps RP, Sime PJ, McBurney MW, Guarente L, Rahman I. SIRT1 protects against cigarette smoke-induced lung oxidative stress via a FOXO3-dependent mechanism. Am J Physiol Lung Cell Mol Physiol 2014; 306(9):L816–L828.
- Zhang T, Rong N, Chen J, Zou C, Jing H, Zhu X, Zhang W. SIRT1 expression is associated with the chemotherapy response and prognosis of patients with advanced NSCLC. PLoS One 2013; 8(11):e79162.
- Chen X, Hokka D, Maniwa Y, Ohbayashi C, Itoh T, Hayashi Y. Sirt1 is a tumor promoter in lung adenocarcinoma. Oncol Lett 2014; 8(1):387–393.
- Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis—the p53 network. J Cell Sci 2003; 116(Pt 20):4077–4085.
- Weaver CV, Liu SP. Differentially expressed pro- and anti-apoptogenic genes in response to benzene exposure: Immunohistochemical localization of p53, Bag, Bad, Bax, Bcl-2, and Bcl-w in lung epithelia. Exp Toxicol Pathol 2008; 59(5):265–272.
- Arunachalam G, Samuel SM, Marei I, Ding H, Triggle CR. Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br J Pharmacol 2014; 171(2):523–535.
- Ganesan S, Unger BL, Comstock AT, Angel KA, Mancuso P, Martinez FJ, Sajjan US. Aberrantly activated EGFR contributes to enhanced IL-8 expression in COPD airways epithelial cells via regulation of nuclear FoxO3A. Thorax 2013; 68(2):131–141.