
Abstract
Desmosomes have long been appreciated as intercellular junctions that are vital for maintaining the structural integrity of stratified epithelia. More recent clinical investigations of patients with diseases such as arrhythmogenic cardiomyopathy have further highlighted the importance of desmosomes in cardiac tissue, where they help to maintain coordination of cardiac myocytes. Here, we review clinical and mechanistic studies that provide insight into the functions of desmosomal proteins in skin and heart during homeostasis and in disease. While intercellular junctions are organized differently in cardiac and epithelial tissues, studies conducted in epithelial systems may inform our understanding of cardiac desmosomes. We explore traditional and non-traditional roles of desmosomal proteins, ranging from adhesive capacities to nuclear functions. Finally, we discuss how these studies can guide future investigations focused on determining the molecular mechanisms by which desmosomal mutations promote the development of cardiac diseases.
INTRODUCTION: CLINICAL ANALYSIS DRAWS ATTENTION TO CARDIAC DESMOSOMES
In 1986, a clinical report emerged describing a group of patients in the Greek island of Naxos with familial palmoplantar keratoderma and hair abnormalities (CitationProtonotarios et al., 1986). Unlike previous reports of epidermal diseases, the majority of these patients were also found to have symptoms or signs of cardiac disease. Patients experienced ventricular tachycardias and were found to have enlarged cardiac ventricles, with one case resulting in sudden cardiac death. In 1996 and 1998, two reports described similar findings of woolly hair, palmoplantar keratoderma, and dilated cardiomyopathy (DCM) in patients in India and Ecuador (CitationCarvajal-Huerta, 1998; CitationRao et al., 1996).
A major step toward understanding molecular mechanisms underlying these cardiocutaneous syndromes, respectively termed Naxos disease and Carvajal syndrome, was taken upon the discovery that many cases were associated with mutations in proteins of intercellular junctions termed desmosomes, reviewed in detail earlier in this series (CitationHarmon & Green, 2013). Naxos disease was mapped to a 2-base-pair deletion in the gene encoding plakoglobin (Pg) causing a truncation of its C-terminus (CitationMcKoy et al., 2000), whereas Carvajal syndrome was mapped to a truncation in the gene encoding desmoplakin (DP) (CitationNorgett et al., 2000).
The emergence of these clinical syndromes sparked a number of reports demonstrating the importance of desmosomal proteins to the structure and function of cardiac tissue. Perhaps most notable are those describing arrhythmogenic cardiomyopathy (AC), a cardiac disease characterized by fibro-fatty replacement of healthy myocardium leading to arrhythmias and in some cases, sudden cardiac death (CitationBasso et al., 2011; CitationHerren et al., 2009). The majority of mutations linked to AC are autosomal dominant and target desmosomal proteins; thus, AC has been dubbed a “disease of the desmosome.” Naxos disease and Carvajal syndrome are considered to be autosomal recessive variants of AC.
While much is yet to be elucidated regarding the roles of desmosomal proteins in the heart, insights can be drawn from studies conducted in epithelial models, where the roles of desmosomes in regulating mechanical integrity and cell signaling are better understood (CitationSimpson et al., 2011). We review those studies here and highlight potential differences between desmosomal proteins in heart and skin that should be accounted for when analyzing the functions of cardiac desmosomes.
CARDIAC DESMOSOMES: A COMPONENT OF THE AREA COMPOSITA
As reviewed earlier in this series (CitationHarmon & Green, 2013), desmosomes are a type of adhesive intercellular junction appearing as electron-dense plaques at regions of cell–cell contact. Desmosomes are composed of proteins from three gene families: cadherins, armadillo proteins, and plakins. Desmosomal cadherins (Desmoglein, Dsg; Desmocollin, Dsc) link desmosomes of opposing cells through their ectodomains, while their cytoplasmic tails connect to desmosomal armadillo proteins (Plakoglobin, Pg; Plakophilin, PKP). These armadillo proteins enable the desmosomal plaque protein desmoplakin (DP) to tether intermediate filaments (IFs) to sites of cell–cell contact (). While complex stratified tissues such as epidermis express multiple cadherin and armadillo isoforms, cardiac tissue isoforms are limited to Dsg2, Dsc2, and PKP2, which along with Pg and DP anchor the IF desmin to the cardiac myocyte plasma membrane (CitationBolling & Jonkman, 2009).
Figure 1. Intercellular junctions in epithelial and cardiac tissues. (A) In epithelial tissues, adherens junctions and desmosomes comprise discrete plaques that respectively tether actin and intermediate filaments to sites of cell–cell contact (CitationHarmon & Green, 2013; CitationSimpson et al., 2011). (B) In cardiac tissue, components of desmosomes and adherens junctions intermingle as a hybrid area composita that stabilizes adjacent gap junctions (CitationBolling & Jonkman, 2009; CitationFranke et al., 2006). Desmoplakin may localize to N-cadherin via plakoglobin (CitationCowin et al., 1986), while αT-catenin could associate with desmosomal cadherins through plakophilin 2 (CitationGoossens et al., 2007). Both desmocollin 2 and plakophilin 2 have been shown to associate with connexin43 (CitationGehmlich et al., 2011a; CitationLi et al., 2009; CitationOxford et al., 2007), potentially forming direct links between the area composita and gap junction plaques.
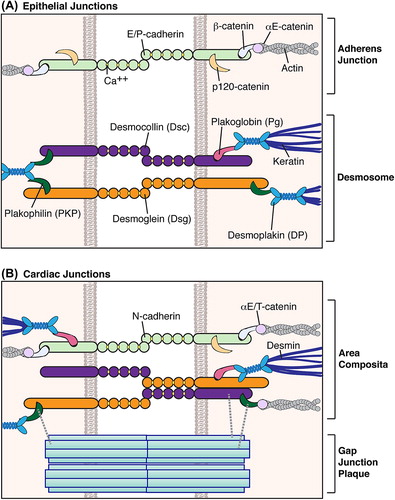
In epithelial cells, desmosomes are discrete structures that tether IF keratins to sites of cell–cell contact, whereas adhesive adherens junctions anchor actin filaments (CitationSimpson et al., 2011). Cardiac desmosomes and adherens junctions, in addition to gap junctions that provide open channels for electrical communication, are traditionally described as discrete components of intercalated discs connecting adjacent cardiac cells. However, more recent studies have revealed that in higher organisms, components of desmosomes and adherens junctions actually intermingle to form a hybrid area composita (CitationFranke et al., 2006) (). This finding necessitates careful consideration of functional differences that may exist for these junctional proteins in different tissue types.
Cardiac muscle architecture
Cardiac tissue is a complex three-dimensional network of cardiac myocytes interspersed with endothelial cells, fibroblasts, and vascular smooth muscle (CitationHarvey & Leinwand, 2011). Cardiac myocytes must maintain mechanical and electrical connections for proper cardiac contraction. Desmosomes and adherens junctions are well known for maintaining structural connections, while gap junctions and ion channels facilitate electrical communication between cells (CitationSimpson et al., 2011).
Disruption of these networks can produce a variety of cardiac diseases (CitationElliott, 2008; CitationHarvey & Leinwand, 2011; CitationMaron et al., 2006). Irregular heartbeats associated with conditions such as arrhythmias can stem from post-ischemic scar tissue, which disrupts electrical coordination of cardiac cell networks. Cardiomyopathies, or diseases of cardiac muscle, are associated with a wide range of inherited or acquired factors (). Many cases of cardiomyopathy are linked to mutations of sarcomeric proteins internal to each cardiac myocyte, which are vital for actin–myosin contraction. AC is associated with mutations in desmosomal proteins, though the extent to which cardiomyopathy arises from a loss of desmosome-based structural coordination or other less understood signaling changes is unclear. The potential bases for AC will be further discussed in this review.
Table 1. Cardiomyopathy (CM) classifications. Generalized clinical findings and etiology of each condition, in addition to those of unclassified cardiomyopathies (left ventricular noncompaction, stress-induced, cirrhotic, etc.) are further described in referenced reviews (CitationElliott, 2008; CitationHarvey & Leinwand, 2011; CitationMaron et al., 2006).
Desmosomes in the heart
From 2006 to 2008, Werner Franke and colleagues published a series of papers describing the discovery of the area composita (CitationBorrmann et al., 2006; CitationFranke et al., 2006, Citation2007; CitationPieperhoff et al., 2008, Citation2010; CitationPieperhoff & Franke, 2007, Citation2008). Previously, it had been thought that desmosomes and adherens junctions represented discrete intercellular junctions, with the exception of Pg having dual localization to both junction types. Upon closer examination, electron microscopy demonstrated that immunogold-labeled desmosomal components (DP, Pg, and PKP2) could localize to over 85% of total cell–cell contact area, including regions resembling adherens junctions (CitationFranke et al., 2006). Thus, desmosomes and adherens junctions are proposed to comprise a hybrid area composita.
Subsequent studies elucidated the timing of area composita development (CitationPieperhoff & Franke, 2007). During early stages of embryogenesis, localization of DP is mutually exclusive with the adherens junction component N-cadherin. These proteins begin to converge at E13, though desmosomal cadherins do not co-localize with N-cadherin until birth. Coalescence of adherens junctions and desmosomes progressively increases from 6 days until 1 year after birth. Non-mammalian vertebrates do not exhibit this mixing of junctions, suggesting that the development of the area composita is a late process of vertebrate evolution (CitationPieperhoff et al., 2010).
Crosstalk between intercellular junctions
A few studies have provided clues as to how desmosomal and adherens junctions components intermingle (). Pg is known to localize to both desmosomal and classical cadherins (CitationCowin et al., 1986), providing a mechanism by which DP could localize to adherens junctions. In fact, Pg has been shown to form an association between DP and VE-cadherin in endothelial cells (CitationKowalczyk et al., 1998). The desmosomal armadillo protein PKP2 interacts with αT-catenin (CitationGoossens et al., 2007), explaining a potential association of actin with desmosomes.
While the area composita in itself refers to a mixing of proteins of adhesive junctions, additional studies have established interactions of these proteins with components of cardiac electrical machinery. Both Dsc2 and PKP2 have been found to co-immunoprecipitate with the gap junction component connexin 43 (Cx43) (CitationGehmlich et al., 2011a; CitationLi et al., 2009; CitationOxford et al., 2007); in fact, PKP2 can be found to localize inside gap junction plaques (CitationAgullo-Pascual et al., 2013). Subsequent studies demonstrated that PKP2 and Dsg2 can associate with and regulate the activity of the cardiac voltage-gated sodium channel Nav1.5 (CitationRizzo et al., 2012; CitationSato et al., 2009). This interplay of desmosomal proteins with electrical machinery may be coordinated by the cytoskeletal adaptor protein ankyrin-G (CitationSato et al., 2011) and could potentially contribute to crosstalk between gap junctions and sodium channel complexes (CitationDesplantez et al., 2012; CitationJansen et al., 2012). These interactions provide a potential explanation for the observed electrophysiological abnormalities in disorders associated with mutations in desmosomal components, including Naxos disease, Carvajal syndrome, and AC (CitationBasso et al., 2011; CitationKaplan et al., 2004a, Citation2004b).
AC: A DISEASE OF THE DESMOSOME
Historical perspective
The first descriptions of AC may have occurred as early as the mid-1700s, when a physician in Rome described a family with palpitations, dilatation and aneurysms of the right ventricle, and sudden death recurring over four generations (CitationLancisi, 1736). The more widely recognized clinical descriptions associated with AC emerged in the late 1970s when Fontaine and colleagues described a condition characterized by replacement of cardiac musculature with fatty and fibrous tissue (CitationFrank et al., 1978; CitationMarcus et al., 1982). At later stages of disease, patients were noted to have enlarged hearts with paper-thin right ventricles. This morphology led clinicians to associate the disease with Uhl's anomaly in which the right ventricular myocardium fails to develop during embryogenesis (CitationUhl, 1952). Due to the belief that the disease was a developmental abnormality, this condition was initially referred to as right ventricular (RV) dysplasia. Fontaine and colleagues proposed a subset of this condition when associated with arrhythmias: arrhythmogenic RV dysplasia (ARVD).
Upon further investigation, however, it was recognized that associated structural defects were not present at birth, but instead represented progressive damage of the myocardium. Together with the growing recognition of arrhythmias as a common finding of the disease, the classification of the condition was changed from a type of dysplasia to a type of cardiomyopathy: arrhythmogenic right ventricular cardiomyopathy (ARVC) (CitationBasso et al., 2009, Citation2011). The World Health Organization recognized ARVC as a distinct class of cardiomyopathy in 1995 (CitationRichardson et al., 1996).
While myocardial damage is most typically found in the right ventricle, involvement of the left ventricle has been observed in as high as half of reported cases (CitationBasso et al., 2009). Left ventricular involvement may be associated with later stages of disease progression (CitationCorrado et al., 1997). However, in many cases morphological changes occur predominantly or solely in the left ventricle (CitationBauce et al., 2005; CitationNorman et al., 2005; CitationOkabe et al., 1995; CitationSen-Chowdhry et al., 2008; CitationSuzuki et al., 2000). While the factors dictating ventricular localization are unknown, these cases have once again prompted a renaming of the disease. Use of ARVC and ARVD are still common, but the disease is increasingly being referred to, broadly, as AC (CitationBasso et al., 2011).
Clinical findings
Patients with AC commonly present in the second to fourth decades of life with syncope, palpitations, and/or arrhythmias resulting in depolarization/repolarization abnormalities on electrocardiogram (CitationBasso et al., 2009). AC more frequently affects men than women. Diagnosis of AC is based on cardiac structural alterations, tissue characterization of ventricular walls, repolarization abnormalities, depolarization/conduction abnormalities, arrhythmias, and family history (CitationMarcus et al., 2010; CitationMcKenna et al., 1994). Athletes with AC are at a higher risk of triggering an arrhythmic event leading to sudden cardiac death, potentially due to acute volume overload and stretching of the right ventricle caused by strenuous activity (CitationBasso et al., 2009; CitationCorrado et al., 2006b).
Compared to other cardiac diseases, AC is considered to be a rare form of cardiomyopathy. Still, AC has a prevalence of up to 1:1000 in certain regions of Europe and 1:5000 in the United States. Further, AC is likely to be under-recognized, remaining unnoticed throughout a patient's life or resulting in sudden cardiac death (CitationBasso et al., 2009, Citation2011; CitationCorrado et al., 2011). This mortality highlights the importance of elucidating disease pathogenesis and improving disease detection/diagnosis. The original AC diagnostic criteria formulated in 1994 were recently revised in an effort to improve sensitivity for detection of early stages and familial forms of AC (CitationMarcus et al., 2010). Screening programs in Italy, in which competitive athletes undergo clinical examination and take prescreening questionnaires to assess the risk of cardiovascular events, have led to a significant decline in sudden cardiac death in athletes aged 12–35 years (CitationCorrado et al., 2006a). The implementation of such programs worldwide will likely result in similar declines in mortality (CitationCorrado et al., 2008), though variable penetrance of AC mutations necessitates more rigorous criteria to determine if it is beneficial to limit sports activity for these patients (CitationCorrado et al., 2006b).
Histologic and molecular characteristics
On histologic examination, AC is associated with replacement of myocardial cells with fatty and fibrous tissue (CitationBasso et al., 2009; CitationHerren et al., 2009; CitationMarcus et al., 1982). This infiltrate typically begins near the epicardium and progressively extends into the endocardium, becoming transmural but leaving the interventricular septum intact. Wall thinning is found to be most severe in the right ventricular “triangle of dysplasia” (anterior infundibulum, right ventricular apex, and inferior aspect of the right ventricle), though the posterolateral left ventricle may be more readily involved than the right ventricular apex (CitationTe Riele et al., 2013). These regions are vulnerable to aneurysm formation from continuous cycles of mechanical stress. While the presence of fatty tissue may be nonspecific, a degenerative change of myocytes with replacement fibrosis is indicative of AC.
AC is hereditary in up to 50% of patients, and studies over the past 15 years have identified mutations in the desmosomal proteins Dsg2, Dsc2, Pg, PKP2, and DP as being responsible for the vast majority of these cases (). Thus, AC is often referred to as a disease of the desmosome (CitationBasso et al., 2009; CitationHerren et al., 2009; CitationRickelt & Pieperhoff, 2012). Mutations in PKP2 appear to be most frequent and are associated with founder groups in regions of the Netherlands (CitationBasso et al., 2009; CitationCox et al., 2011). However, mutations in any of these proteins may be non-pathogenic polymorphisms, and variable disease penetrance may impede predictions of phenotypic outcome based on genetics. The online ARVC database (http://www.arvcdatabase.info) is a valuable tool that uses in silico prediction models and clinical data to assess mutation pathogenicity (Citationvan der Zwaag et al., 2009). Additionally, a study recently identified the N-terminal regions of DP and Dsg2, in addition to conserved residues of PKP2 and Dsg2, as “hot spots” for radical mutations associated with AC development (CitationKapplinger et al., 2011). However, little is known regarding the mechanisms by which these mutations promote disease pathogenesis.
Table 2. Mutations associated with AC and AC-overlap syndromes. Detailed descriptions of the listed mutations can be found in the literature cited in this text and elsewhere (CitationRickelt & Pieperhoff, 2012; Citationvan der Zwaag et al., 2009).
Mutations in other non-desmosomal proteins have also been associated with AC development, though they are less common than desmosomal mutations (). For example, mutations have been found in transforming growth factor beta 3 (TGF-β3) (CitationBao et al., 2013; CitationBeffagna et al., 2005). TGF-β3 is a key regulator of fibrotic signaling pathways (CitationLeask & Abraham, 2004; CitationVarga & Jimenez, 1986); thus, these mutations could induce fibrosis by stimulating proliferation of mesenchymal cells. Alternatively, TGFB3 mutations could act by altering expression of Pg (CitationKapoun et al., 2004) and DP (CitationYoshida et al., 1992), supporting a theory that non-desmosomal mutations can still act through misregulation of desmosomes.
Following this theory, AC was recently associated with a mutation in the adherens junction component αT-catenin (Citationvan Hengel et al., 2013). αT-catenin knockout mouse models exhibit a redistribution of PKP2 (CitationLi et al., 2012b). As αT-catenin can associate with PKPs (CitationGoossens et al., 2007; Citationvan Hengel et al., 2013), it is plausible that αT-catenin mutations in AC ultimately act through misregulation of desmosomal proteins.
Screening of AC patients revealed a founder mutation in phospholamban, a regulator of the sarcoplasmic reticulum Ca2+ (SERCA2a) pump in cardiac muscle (CitationGroeneweg et al., 2013; Citationvan der Zwaag et al., 2012, Citation2013). Interestingly, SERCA2 regulates desmosomal adhesive strength by promoting membrane localization of DP (CitationHobbs et al., 2011), once again raising the possibility that desmosomal misregulation is a key step in AC development.
A cohort of patients in Newfoundland, Canada expresses a fully penetrant, lethal version of AC associated with a mutation targeting TMEM43, which encodes the inner nuclear membrane protein LUMA (CitationMerner et al., 2008). TMEM43 mutations were subsequently identified in patients outside of this cohort (CitationBao et al., 2013; CitationBaskin et al., 2013; CitationChristensen et al., 2011; CitationHaywood et al., 2013; CitationLarsen et al., 2012). TMEM43 has been proposed to be part of the adipogenic peroxisome proliferator-activated receptor γ (PPARγ) pathway (CitationMerner et al., 2008), suggesting that mutations may hyperactivate adipogenesis. LUMA binds lamins and emerins to contribute to nuclear envelope integrity (CitationBengtsson & Otto, 2008), and a mutation in lamin A/C has recently been associated with AC (CitationQuarta et al., 2012). A potential relationship between these mutations and desmosomal misregulation has not been established.
Mutations in the sarcomeric protein titin (TTN) (CitationTaylor et al., 2011) and the IF desmin (DES) (CitationLorenzon et al., 2013; CitationHedberg et al., 2012; CitationKlauke et al., 2010; Citationvan Tintelen et al., 2009) have been associated with AC but are sometimes considered to produce phenocopies or AC-overlap syndromes (CitationBasso et al., 2011; CitationMcLendon & Robbins, 2011; CitationOtten et al., 2010; Citationvan Spaendonck-Zwarts et al., 2011). It is likely that future studies will reveal additional non-desmosomal mutations associated with AC. Further screening of large patient cohorts is needed to assess the prevalence of these mutations in AC patients, to determine the specificity of mutations to AC versus other cardiac diseases, and to elucidate if and how their pathogenicities relate to desmosomal misregulation.
Theories of AC pathogenesis
Though histological findings of replacement-type fibrosis, fat deposition, and myocyte degeneration are hallmarks of AC, the mechanisms by which these changes arise are poorly understood. Possible causes are grouped into three theories of pathogenesis lacking mutual exclusivity: degeneration, inflammation, and transdifferentiation () (CitationBasso et al., 2011; CitationHerren et al., 2009).
Figure 2. Theories of AC pathogenesis. AC may arise from cardiac myocyte degeneration, inflammation of myocardium from infection by cardiotropic pathogens, and/or transdifferentiation of cardiac myocytes or progenitor cells into fat- and fibrotic scar-producing cells (CitationBasso et al., 2011; CitationHerren et al., 2009).
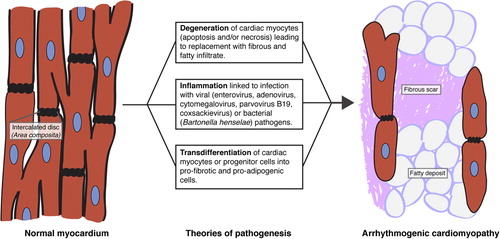
In the degenerative theory, it is proposed that cardiac myocyte death enables replacement with fibrous and fatty tissue. A number of studies have pointed to apoptotic death (CitationCampian et al., 2011; CitationGarcia-Gras et al., 2006; CitationNagata et al., 2000; CitationNishikawa et al., 1999; CitationValente et al., 1998; CitationYamaji et al., 2005). Supporting this idea, fragmented DNA and expression of protease CPP-32 were present in 75% of myocardial samples in one study of AC patients (CitationMallat et al., 1996), and expression of an AC PKP2 mutation in cardiac cells increased levels of apoptosis (CitationJoshi-Mukherjee et al., 2008). However, there are also examples of necrosis in human samples and animal models of AC (CitationGattenlohner et al., 2009; CitationLi et al., 2011b; CitationPilichou et al., 2009). It is plausible that more than one cell death pathway is involved in AC development (CitationLi et al., 2011a).
Inflammatory infiltrates are commonly found in AC, suggesting that inflammation could contribute to AC development (CitationAsimaki et al., 2011; CitationBasso et al., 1996; CitationCalabrese et al., 2006; CitationFornes et al., 1998). Non-familial AC may be linked to inflammation from infections by viruses including enterovirus, adenovirus, cytomegalovirus, parvovirus B19, and coxsackievirus (CitationBowles et al., 2002; CitationGrumbach et al., 1998), or by the bacterium Bartonella henselae (CitationH Fischer et al., 2008). It has been proposed that this inflammation can be noninvasively detected by assaying plasma levels of IL-1β, IL-6, and TNF-α (CitationCampian et al., 2010). However, AC patients sometimes have very little evidence of inflammation (CitationTabib et al., 2003). Inflammatory infiltrate could be secondary to cell death, or could be limited to severe biventricular forms of AC (CitationCampuzano et al., 1986). Thus, the source of inflammation and the value of its assessment remain unclear.
Finally, observations of myocardial cells expressing altered cytological markers have led to the development of a transdifferentiation theory. Since adult cardiac myocytes have limited capabilities for dedifferentiation, it is possible that pro-adipogenic cells found in AC samples are derived from progenitor cells in the subepicardial or second heart fields (CitationGittenberger-de Groot et al., 2010; CitationLombardi et al., 2009; CitationMatthes et al., 2011). Multiple AC studies have noted an accumulation of fat droplets or activation of adipogenic pathways in cardiac myocytes (CitationChen et al., 2014; CitationD’Amati et al., 2000; CitationFujita et al., 2008; CitationGarcia-Gras et al., 2006; CitationKim et al., 2013), though it is unclear if these changes are indicative of transdifferentiation.
There is much to be understood regarding the specific mechanisms by which desmosomal mutations may trigger these pathways of disease pathogenesis. Recent work has started to evaluate how loss of desmosomal adhesion and/or signaling promotes cell death, increases susceptibility for viral infection, and/or promotes fibro-fatty change. These pathways will be eplained later in this review.
Desmosomal misregulation in other cardiac diseases
Clinical reports have demonstrated that desmosomal proteins are mutated or otherwise misregulated in a broader spectrum of cardiac diseases. A growing number of studies have established links between desmosomal proteins and components of cardiac electrical machinery including the gap junction subunit connexin43 (Cx43) (CitationAgullo-Pascual et al., 2013; CitationGehmlich et al., 2011a; CitationOxford et al., 2007; CitationSato et al., 2011) and the sodium channel isoform Nav1.5 (CitationCerrone et al., 2012; CitationRizzo et al., 2012; CitationSato et al., 2009). Therefore, it is possible that desmosomal mutations may contribute more generally to cardiac diseases involving arrhythmia. For example, mutations in PKP2 proposed to cause Nav1.5 mislocalization were recently described in patients with Brugada syndrome (CitationCerrone et al., 2013). Similarly, mutations in PKP2 have been associated with sudden unexplained death with negative autopsy (SUDNA) in which cardiac tissue is morphologically normal and therefore not definitively marked as AC (CitationZhang et al., 2012).
PKP2 was also found to be a marker of cardiac myxomata derived from mesenchymal cells (CitationRickelt et al., 2010). PKP2 rapidly accumulates in abnormal adherens junction complexes in myxoma samples, without accumulation of other desmosomal proteins. The mechanisms by which PKP2 localizes to adherens junctions in these samples, and the significance of this finding as being a cause versus marker of disease, are yet to be determined.
Desmosomal mutations have also been associated with DCM (CitationElliott et al., 2010; CitationPugh et al., 2014). One study noted that 13% of DCM patients harbored desmosomal mutations predicted to be pathogenic, and an additional 6% had desmosomal mutations of unknown significance (CitationGarcia-Pavia et al., 2011).
The IF desmin is a vital cytoskeletal component of striated muscle, and desmin mutations have been associated with DCM (CitationLi et al., 1999) and with a more generalized “desmin myopathy” characterized by skeletal muscle weakness, arrhythmias, and restrictive heart failure (CitationGoldfarb et al., 2004). Desmin myopathy can be caused by mutations in the desmin rod or tail domains, and is typically associated with intracytoplasmic desmin-reactive deposits (CitationGoldfarb et al., 1998; CitationDalakas et al., 2003).
Finally, while the loss of Pg from intercalated discs has been proposed to be prognostic for AC (CitationAsimaki et al., 2009), a reduction in junctional Pg was also noted in granulomatous myocarditis and sarcoidosis (CitationAsimaki et al., 2011). Pathogenesis of these diseases may be linked to increased cytokine signaling (CitationAsimaki et al., 2011) and/or activation of adipogenic and fibrotic pathways (CitationGarcia-Gras et al., 2006), as can occur upon mislocalization of Pg.
The spectrum of conditions associated with desmosomal misregulation highlights the need to better understand how desmosomal mutations are a basis for disease development.
DESMOSOMES IN DISEASE: MECHANISTIC INSIGHTS
Biochemical analyses, molecular biology, and physiological studies have revealed non-traditional roles for desmosomal proteins that may provide insight to understanding cardiac disease pathogenesis. We review those studies here, highlighting mouse models and studies of individual disease mutations (). We refer readers to other reviews for additional information on the basic biology of desmosomes (CitationDelva et al., 2009; CitationHarmon & Green, 2013) and their roles in human disease (CitationDelmar & McKenna, 2010; CitationMcGrath, 1999).
Table 3. Potential mechanisms of AC mutation pathogenicity. Select findings from mouse models and/or in vitro studies are highlighted here, with further information available in the text.
Desmosomal cadherins
Similar to classical cadherins of adherens junctions, the extracellular domains of desmosomal cadherins form adhesive bonds between desmosomal plaques of neighboring cells. As such, mutation or autoimmune-mediated targeting of desmosomal cadherins can lead to blistering disorders including pemphigus vulgaris, pemphigus foliaceus, and keratodermas, reviewed elsewhere (CitationAmagai, 2010; CitationGetsios et al., 2010; CitationWaschke, 2008). Misregulation of desmosomal cadherins has also been associated with defects in hair growth/cycling (CitationMcGrath & Wessagowit, 2005). Here, we will focus on misregulation of desmosomal cadherins expressed in cardiac tissue: Dsg2 and Dsc2.
Dsg
Dsg2-knockout mouse models demonstrate a vital function for Dsg2 in development. Dsg2 is needed for embryonic stem cell proliferation, and Dsg2-/- mice die at or shortly after implantation (CitationEshkind et al., 2002). More restricted targeting of Dsg2 through transgenic expression of Dsg2 N271S, the mouse equivalent of the human AC mutation Dsg2 N266S, reproduces features of AC with myocyte necrosis thought to initiate myocardial damage (CitationPilichou et al., 2009). Mice expressing a Dsg2 mutant lacking the Dsg2 ectodomain demonstrate cell death and fibrosis associated with increased mRNA expression of cardiac stress and remodeling markers, including c-Myc, ANF, BNF, CTGF, and GDF15 (CitationKrusche et al., 2011). Importantly, necrosis in Dsg2 mutant models is preceded by widening of intercellular spaces with abnormalities in ventricular activation time, attributed to the novel finding of an interaction between Dsg2 and the sodium channel Nav1.5 (CitationRizzo et al., 2012). Thus, necrosis may be a secondary, late stage of disease following more specific signaling changes.
Alterations in molecular pathways that may contribute to disease pathogenesis induced by expression of Dsg2 mutations have been revealed by recent biochemical analyses. AC mutation C813R targeting the intracellular cadherin segment (ICS) domain of Dsg2 causes a shift in protein migration detected by gel electrophoresis (CitationGehmlich et al., 2010). Migration patterns were restored by the mutant C813A, suggesting that mutations may introduce polar residues that alter posttranslational modifications of Dsg2. The V977fsX1006 frameshift mutation in the Dsg2 unique region (DUR) of Dsg2 compromised tail–tail interactions of homodimerized Dsg2, leading to an increased rate of Dsg2 endocytosis and thus loss of cell surface Dsg2 (CitationChen et al., 2012). Further, the R46Q mutation in the proprotein convertase cleavage site of Dsg2 impairs proper prodomain cleavage, prerequisite to normal Dsg2 function in desmosomes (CitationGaertner et al., 2012). Because it has been observed that mutated Dsg2 proteins can be incorporated into desmosomes (CitationRasmussen et al., 2013b), it is likely that phenotypes induced by these mutations promote disease development even when co-expressed with wild-type Dsg2.
Interestingly, Dsg2 has been identified as a high-affinity receptor used by multiple adenovirus serotypes (CitationWang et al., 2011a,Citationb, Citation2013). More information is needed to establish if and how mutated Dsg2 may enhance AC development upon viral infection.
Dsc
Similar to some Dsg2 mutations, missense mutations R203C and T275M in Dsc2 hinder proteolytic cleavage of the Dsc2 prodomain, causing a reduction in functional Dsc2 (CitationGehmlich et al., 2011b). Missense mutations E102K targeting the Dsc2 prodomain and I345T targeting the Dsc2 ectodomain result in cytoplasmic localization of Dsc2, likely by respectively impairing proteolytic cleavage and adhesive functions of Dsc2 (CitationBeffagna et al., 2007). Interestingly, a recent study found that the Dsc2a splice variant can directly interact with the gap junction component Cx43, and that expression of the mutation Q851fsX855 compromises binding of Dsc2a with both Cx43 and Pg (CitationGehmlich et al., 2011a). Thus, mutations in desmosomal cadherins can impair mechanical and electrical coordination of cardiac cells through distinct mechanisms.
Armadillo proteins
The structural similarities between desmosomal armadillo proteins (Pg and PKPs) and their respective counterparts in adherens junctions (β-catenin and p120 catenin) have prompted analysis of the unique and overlapping functions of these junctional proteins (CitationHatzfeld, 2007; CitationBass-Zubek et al., 2009; CitationZhurinsky et al., 2000b). Armadillo proteins not only play structural roles in junctions but also activate and repress transcription of nuclear genes (CitationMiller et al., 2013; CitationSchmidt & Koch, 2007). The extent to which their junctional and nonjunctional functions contribute to human disease pathogenesis is yet to be fully determined.
Pg
Pg (also known as γ-catenin) is homologous to the adherens junction component β-catenin (CitationButz et al., 1992; CitationMcCrea et al., 1991), an effector of the canonical Wnt signaling pathway that promotes T-cell/lymphoid-enhancing binding (TCF/LEF) transcriptional activity. Pg-null mouse models demonstrate embryonic lethality between E12 and 16 with a “broken heart” phenotype: cardiac ventricles burst and blood fills the pericardium (CitationRuiz et al., 1996). The architecture of intercellular junctions is severely altered in these mice, with a diffuse localization of Dsg2, abnormally elongated junctions, and an enhanced co-localization of DP with β-catenin at early time points of development. β-catenin can partially compensate for the absence of Pg in the skin, where it associates with desmosomal cadherins and reduces the severity of skin phenotypes in Pg-null mice (CitationBierkamp et al., 1999; CitationLi et al., 2012a).
Importantly, while Pg and β-catenin may replace each other in junctions, their structural similarities may actually lead to functional competition in the nucleus (CitationBen-Ze'ev & Geiger, 1998; CitationHu et al., 2003; CitationSimcha et al., 1998; CitationZhurinsky et al., 2000a) since the arm domain mediates binding of both proteins to TCF/LEF nuclear partners (CitationBehrens et al., 1996; CitationHuber et al., 1996; CitationSimcha et al., 1998; CitationMiravet et al., 2002). For example, cardiac-restricted deletion of Pg leads to a concomitant increase in β-catenin at junctions and in the nucleus, where it associates with TCF-4 to activate transcription of factors including c-Myc, c-Fos, and cyclin D1 (CitationLi et al., 2011b). In contrast, Pg has been shown to form a complex with LEF-1 to repress transcription of c-Myc in the epidermis (CitationWilliamson et al., 2006).
Interestingly, cardiac deletion of DP results in relocalization of Pg to the nucleus, where it suppresses Wnt/β-catenin signaling and activates expression of adipogenic and fibrotic genes including C/EBP-α, PPARγ, adiponectin, lipoprotein lipase, and procollagens (CitationGarcia-Gras et al., 2006). However, a separate study of Pg ablation in cardiac myocytes revealed an AC phenocopy with increased cell death but without a change in Wnt/β-catenin-mediated signaling (CitationLi et al., 2011a). Instead, these mice were found to have elevated levels of TGF-β-mediated signaling. Thus, mouse models have not fully clarified mechanisms by which Pg and β-catenin are regulated, or by which they may affect each other's activities (CitationZhurinsky et al., 2000b).
Since a Pg mutation associated with Naxos disease (Pg2157del2) produces a truncated form of Pg that is still detectable in patient biopsies (CitationMcKoy et al., 2000), further information may be gained by studying mutated Pg rather than Pg-null models. C-terminal truncation mutants of Pg can be incorporated into desmosomes but alter junction morphology and produce extremely long desmosomes (CitationPalka & Green, 1997). A transgenic mouse model overexpressing truncated Pg confirmed a loss of binding to DP and Dsg2 (CitationLombardi et al., 2011). As truncated Pg localizes primarily to the nucleus, cardiac progenitor cells isolated from these mice demonstrate increased levels of adipogenic factors including KLF15, C/EBP-α, and Wnt5β, and reduced levels of the anti-adipogenic factor CTGF. Thus, these more specific mutant models are useful in determining mechanisms of disease pathogenesis.
Expression of an N-terminal S39_K40insS Pg mutation associated with AC has demonstrated increased ubiquitylation and cytoplasmic localization of Pg (CitationAsimaki et al., 2007). Further, this Pg mutant revealed novel interactions with histidine-rich calcium-binding protein (HRC-BP) and TGF-β-induced apoptosis protein 2 (TAIP-2), producing higher rates of proliferation and lower rates of apoptosis. While expression of either Naxos-associated Pg2157del2 or S39_K470insS increases cell motility in epithelial cells, 2157del2 does so by compromising cell–cell adhesion while S39_K470insS decreases cell stiffness (CitationHuang et al., 2008). In vitro studies have demonstrated that Pg can regulate fibronectin production and Src signaling via the small GTPases Rac1 and RhoA in epithelial cells (CitationYin et al., 2005; CitationTodorovic et al., 2010). Together, these observations suggest that Pg mutations could act by impairing desmosomal and non-desmosomal functions.
PKP
PKPs 1, 2, and 3 have variable expression patterns in different tissues: PKP1 is most highly expressed in suprabasal layers of stratified epithelia, PKP2 is expressed in all simple and complex epithelia in addition to cardiac myocytes and lymph node follicle cells, and PKP3 is expressed in most simple epithelia in addition to all layers of stratified epithelia (CitationBass-Zubek et al., 2009; CitationHatzfeld, 2007). PKPs localize to desmosomes in addition to cell nuclei, with this differential localization potentially dictated by alternative splice products (CitationSchmidt et al., 1997) or by the interaction of PKPs with scaffolding proteins such as 14-3-3 (CitationMuller et al., 2003).
While a potential role for PKPs in controlling gene transcription is uncertain, it is clear that they have functions in addition to their roles in adhesion. PKP2 can interact with the RNA polymerase III subunit of the transcription factor TFIIIB (CitationMertens et al., 2001) and can augment β-catenin/TCF-mediated transcription (CitationChen et al., 2002). PKP3 forms cytoplasmic stress-induced granules with RNA-binding proteins PABPC1, FXR1, and G3BP (CitationHofmann et al., 2006), and has recently been shown to associate with the ETV1/ER81 transcription factor (CitationMunoz et al., 2014). While all PKPs may be important in the pathogenesis of human diseases including ectodermal dysplasia-skin fragility syndrome (CitationMcGrath & Wessagowit, 2005), here we will focus on studies of PKP2, the sole PKP expressed in heart.
PKP2-null mice lack obvious epithelial defects, likely due to compensation from other PKP isoforms; yet, these mice experience embryonic lethality at E10.5–E11 due to severe alterations in cardiac morphology (CitationGrossmann et al., 2004). Mice with PKP2 haploinsufficiency are viable but exhibit sodium current deficiencies (CitationCerrone et al., 2012), supporting data on the association of PKP2 with the Nav1.5 channel in cells (CitationSato et al., 2009, Citation2011). In cardiac cell cultures, PKP2 knockdown compromises gap junction signaling and Cx43 localization to sites of cell–cell contact (CitationOxford et al., 2007; CitationFidler et al., 2009). In fact, the cytoskeletal adaptor protein ankyrin-G may coordinate interactions between PKP2, Cx43, and Nav1.5 (CitationSato et al., 2011).
In epithelial cells, PKP2 was shown to regulate the localization of RhoA to sites of cell–cell contact, with PKP2 deficiency resulting in an impairment of cortical actin remodeling, an increase in contractile signaling, and alterations in focal contact structure and integrin expression (CitationGodsel et al., 2010; CitationKoetsier et al., 2014). Similar alterations in actin have been observed in PKP2-deficient HL-1 and neonatal rat cardiac myocytes (Dubash et al., unpublished observations). This work raises the possibility that PKP2 could control matrix remodeling in AC. Further, PKP2 forms a complex with DP and PKCα to regulate the recruitment of DP to cell–cell borders (CitationBass-Zubek et al., 2008). PKCα activity is thought to be responsible for a switch between dynamic and “hyperadhesive” states of desmosomes (CitationWallis et al., 2000; Citationvan Hengel et al., 1997), suggesting that misregulation of PKCα localization and/or activity could compromise structural integrity of cell networks including that of cardiac muscle. Thus, misregulation of PKP2 could act in part by affecting its downstream regulation of the desmosomal plaque protein, DP. Further, PKP2-knockdown cardiac cells exhibit reduced levels of active PKCα with subsequent hyperactivation of the Hippo pathway, leading to enhanced adipogenesis that likely contributes to AC (CitationChen et al., 2014).
Some PKP2 mutations are thought to result in loss of protein: for example, the PKP2 mutant c.2386T> C exhibits enhanced degradation by calpain proteases (CitationKirchner et al., 2012). However, other studies have demonstrated that PKP2 mutations can result in stable, dysfunctional PKP2 (Citationvan der Smagt et al., 2012), highlighting the importance of studying both knockdown and mutant PKP2 models. With recent advances in stem cell biology, researchers have begun to explore the pathogenicities of specific mutations using induced pluripotent stem cell (iPSC)-derived cardiac myocytes grown from fibroblasts of patients with PKP2 mutations (CitationMa et al., 2013). iPSC-derived cardiac myocytes expressing the PKP2 mutation c.1841T> C demonstrate an accumulation of lipid droplets. Similarly, cells derived from a patient with the PKP2 c.2484C> T mutation exhibit nuclear translocation of Pg with decreased β-catenin activity (CitationKim et al., 2013). When altering metabolic conditions in culture by modulating PPAR-γ activity, these cells reproduce aspects of AC in vitro with increased lipogenesis and apoptosis. Application of an inhibitor of glycogen synthase kinase 3β (GSK3β) prevents adipogenesis in iPSC-derived cardiac myocytes expressing the c.972InsT/N PKP2 mutation (CitationCaspi et al., 2013), suggesting a potential therapeutic strategy for patients with AC.
DP
DP is an obligate component of desmosomes vital for integrity of multiple tissue types including the skin and heart. DP ablation leads to embryonic lethality by E6.5 with defects in extraembryonic tissue integrity (CitationGallicano et al., 1998). Extra-embryonic rescue of DP via a tetraploid aggregation method delays lethality to E12.5, with cardiac hemorrhages and defective capillary formation (CitationGallicano et al., 2001). Conditional depletion of DP from the epidermis enables embryonic survival but leads to death soon after birth due to severe sloughing of skin (CitationVasioukhin et al., 2001). Interestingly, while adherens junctions are typically thought to regulate desmosome assembly, conditional DP-knockout mice demonstrate abnormalities in adherens junctions. Thus, the assembly, maturation, and maintenance of desmosomes and adherens junctions may be more interdependent than previously recognized. Conditional ablation of DP from cardiac tissue recapitulates AC with defects in connexin expression (CitationLyon et al., 2013) and electrophysiological abnormalities before the onset of overt structural changes (CitationGomes et al., 2012). Changes in gap junction and sodium channel signaling upon DP knockdown have been reproduced in cardiac cell systems (CitationZhang et al., 2013).
In humans, expression of a truncating mutation in the DP C-terminus completely ablates the function of DP in tethering IFs to desmosomes, resulting in the fatal condition of lethal acantholytic epidermolysis bullosa (CitationBolling et al., 2010; CitationHobbs et al., 2010; CitationJonkman et al., 2005). DP mutations and haploinsufficiency have also been associated with less severe epidermal conditions including skin fragility/woolly hair syndrome, hypo/oligodontia, and striate palmoplantar keratoderma, some of which can be associated with cardiac findings (CitationArmstrong et al., 1999; CitationChalabreysse et al., 2011; CitationMahoney et al., 2010; CitationSmith et al., 2012; CitationWhittock et al., 1999, Citation2002).
Biological analysis of individual DP mutations is needed to fully understand the pathogenesis of AC and these epidermal diseases. AC mutations V30M and Q90R targeting the N-terminus of DP display an inability to bind Pg in cells; however, transgenic expression of these mutations leads to embryonic lethality, preventing further understanding of how these mutations might contribute to gradual AC development (CitationYang et al., 2006). In this same study, transgenic expression of R2834H targeting the C-terminus of DP recapitulates AC findings by 6 months of age with lipogenesis, fibrosis, and an absence of desmin at intercalated discs. These mice also demonstrate an increase in cell apoptosis; however, a separate study has shown that RNAi-mediated silencing of DP in keratinocytes actually increases cell proliferation (CitationWan et al., 2007).
It is possible that the domain of DP targeted by AC mutations may dictate pathogenic effects and potential lethality. Mutations targeting the N-terminus of DP could completely prevent localization of DP to desmosomes, whereas mutations targeting the DP C-terminus could allow for partial function of DP in desmosomal plaques but impair tethering of IFs to sites of cell–cell contact. AC mutations may also have different effects on DP expression levels: one study demonstrated that mutant proteins could either be expressed in cells or lead to degradation/instability producing functional haploinsufficiency (R1269X, K324_E325del) (CitationRasmussen et al., 2013a).
There is much to be understood regarding novel functions of DP that stand apart from its tethering of IFs. As mentioned, DP knockout mice activate adipogenesis and fibrosis via nuclear translocation of Pg, antagonistic of canonical Wnt/β-catenin signaling (CitationGarcia-Gras et al., 2006). In support of this finding, DP was shown to act as a tumor suppressor by inhibiting Wnt/β-catenin signaling in a model of non-small cell lung cancer (CitationYang et al., 2012). Recent studies also implicate DP in binding the microtubule plus-end binding protein EB1 to promote Cx43 membrane localization (Patel and Green, in revision). These findings highlight potential novel mechanisms by which DP mutations can contribute to AC development.
IFs
Misregulation of desmosome-associated IFs is implicated in a number of diseases (CitationOmary, 2009). It is unclear if mutations in desmin, the IF expressed in cardiac muscle, primarily cause AC (CitationLorenzon et al., 2013; CitationKlauke et al., 2010; CitationHedberg et al., 2012) or AC phenocopies (Citationvan Tintelen et al., 2009; Citationvan Spaendonck-Zwarts et al., 2011; CitationMcLendon & Robbins, 2011; CitationBasso et al., 2011). Similar to broad findings of desmin myopathy, desmin-null mice develop defects in multiple muscle types (CitationMilner et al., 1996; CitationLi et al., 1996). The determinants of desmin misregulation affecting multiple tissue types as opposed to cardiac muscle alone are not well understood; nonetheless, some valuable information has emerged regarding the pathogenicities of desmin mutations (CitationBar et al., 2004; CitationClemen et al., 2013).
Several desmin mutations located in the central α-helical rod domain of desmin affect filament formation (CitationBrodehl et al., 2013; CitationBar et al., 2005; CitationDagvadorj et al., 2003; CitationSjoberg et al., 1999; CitationMunoz-Marmol et al., 1998; CitationGoldfarb et al., 1998; CitationGoudeau et al., 2001) or can compromise association of desmin with nebulin, through which it attaches to cardiac Z-discs (CitationConover & Gregorio, 2011). Interestingly, disease mutations have been reported to target putative PKC sites in the desmin head domain (CitationPica et al., 2008), a region whose phosphorylation is proposed to regulate cardiac myocyte differentiation (CitationHollrigl et al., 2007).
Truncation of the tail domain of desmin can abolish interactions with DP (CitationLapouge et al., 2006). Mice transgenically expressing the I451M mutation targeting the desmin tail demonstrate a loss of desmin localization to Z-discs (CitationMavroidis et al., 2008). Though desmin I451M can still localize to intercalated discs, junctions exhibit altered morphology. Interestingly, expression of the desmin mutation R454W compromises junctional localization of PKP2 and DP (CitationOtten et al., 2010). Further, a deletion mutation in desmin has been shown to impair membrane localization of Cx43 (CitationGard et al., 2005). Thus, though IFs are an “end-point” of desmosomes, it is important to consider them as potential regulators of junction assembly and stability. More information on these pathways is likely to be obtained from the use of iPSC-derived cardiac myocytes from patients with desmin mutations (CitationTse et al., 2013).
CONCLUDING REMARKS
Historically, desmosomes have been considered as rigid plaques that maintain adhesive integrity of cells in contact. However, as outlined here, the dynamic natures of desmosomal components and their novel non-desmosomal roles are becoming increasingly recognized as key determinants of tissue homeostasis and disease pathogenesis. Researchers and clinicians are able to draw from studies conducted in vitro and in vivo to understand how mutations in these desmosomal proteins can misregulate various cellular processes.
Since the discovery of AC as a “disease of the desmosome,” many patients with cardiac findings have been screened for mutations in desmosomal proteins. However, there are unfortunately a number of confounding factors complicating this approach. Most obviously, the variable penetrance of mutations is poorly understood and may be related to exercise-induced stress, gene–dose effects, and compound and/or digenic heterozygosity (CitationBasso et al., 2011; CitationCorrado et al., 1997, Citation2006b; CitationKirchhof et al., 2006; CitationRigato et al., 2013; CitationXu et al., 2010). Systems analysis may be needed to better determine if AC-associated mutations are non-pathogenic, disease modifying, or highly pathogenic (Citationvan der Zwaag et al., 2009; CitationSilvano et al., 2013; CitationKapplinger et al., 2011; CitationChristensen et al., 2010). Traditional screening methods may lack sensitivity for copy number variations or cryptic splice site mutations in desmosomal genes (CitationAwad et al., 2006; CitationLi Mura et al., 2013), and often neglect consideration of mutations in lesser-studied genes.
Despite these issues, studies analyzing the pathogenicities of individual mutations are essential for gaining a better understanding of disease development. A number of studies have used structural modeling to predict potential mechanisms by which AC mutations could alter protein function (CitationAl-Jassar et al., 2013; CitationChoi & Weis, 2011), though very few studies have expressed these mutations to analyze their effects in vitro or in vivo. Use of iPSC-derived cardiac myocytes obtained from patients with AC mutations, with consideration of epigenetic factors that may contribute to disease development, is a specific method to test misregulation of adhesive and signaling pathways. In the long-term, an integration of molecular modeling, in vitro analysis, and iPSC-based biology may provide key information regarding mechanisms by which desmosomal mutations serve as a basis for disease development.
Declaration of interest: The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Agullo-Pascual E, Reid DA, Keegan S, Sidhu M, Fenyo D, Rothenberg E, Delmar M (2013). Super-resolution fluorescence microscopy of the cardiac connexome reveals plakophilin-2 inside the connexin43 plaque. Cardiovasc Res. 100: 231–240.
- Al-Jassar C, Bikker H, Overduin M, Chidgey M (2013). Mechanistic basis of desmosome-targeted diseases. J Mol Biol. 425: 4006–4022.
- Amagai M (2010). Autoimmune and infectious skin diseases that target desmogleins. Proc Jpn Acad Ser B Phys Biol Sci. 86: 524–537.
- Armstrong DK, McKenna KE, Purkis PE, Green KJ, Eady RA, Leigh IM, Hughes AE (1999). Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum Mol Genet. 8: 143–148.
- Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, McKenna WJ (2007). A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 81: 964–973.
- Asimaki A, Tandri H, Duffy ER, Winterfield JR, Mackey-Bojack S, Picken MM, Cooper LT, Wilber DJ, Marcus FI, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, Stevenson WG, McKenna WJ, Gautam S, Remick DG, Calkins H, Saffitz JE (2011). Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 4: 743–752.
- Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, McKenna WJ, Calkins H, Saffitz JE (2009). A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 360: 1075–1084.
- Awad MM, Dalal D, Tichnell C, James C, Tucker A, Abraham T, Spevak PJ, Calkins H, Judge DP (2006). Recessive arrhythmogenic right ventricular dysplasia due to novel cryptic splice mutation in PKP2. Hum Mutat. 27: 1157.
- Bao JR, Wang JZ, Yao Y, Wang YL, Fan XH, Sun K, Zhang S, Hui RT, Song L (2013). Screening of pathogenic genes in Chinese patients with arrhythmogenic right ventricular cardiomyopathy. Chin Med J (Engl). 126: 4238–4241.
- Bar H, Fischer D, Goudeau B, Kley RA, Clemen CS, Vicart P, Herrmann H, Vorgerd M, Schroder R (2005). Pathogenic effects of a novel heterozygous R350P desmin mutation on the assembly of desmin intermediate filaments in vivo and in vitro. Hum Mol Genet. 14: 1251–1260.
- Bar H, Strelkov SV, Sjoberg G, Aebi U, Herrmann H (2004). The biology of desmin filaments: how do mutations affect their structure, assembly, and organisation?J Struct Biol. 148: 137–152.
- Baskin B, Skinner JR, Sanatani S, Terespolsky D, Krahn AD, Ray PN, Scherer SW, Hamilton RM (2013). TMEM43 mutations associated with arrhythmogenic right ventricular cardiomyopathy in non-Newfoundland populations. Hum Genet. 132: 1245–1252.
- Bass-Zubek AE, Godsel LM, Delmar M, Green KJ (2009). Plakophilins: multifunctional scaffolds for adhesion and signaling. Curr Opin Cell Biol. 21: 708–716.
- Bass-Zubek AE, Hobbs RP, Amargo EV, Garcia NJ, Hsieh SN, Chen X, Wahl JK, 3rd, Denning MF, Green KJ (2008). Plakophilin 2: a critical scaffold for PKC alpha that regulates intercellular junction assembly. J Cell Biol. 181: 605–613.
- Basso C, Bauce B, Corrado D, Thiene G (2011). Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 9: 223–233.
- Basso C, Corrado D, Marcus FI, Nava A, Thiene G (2009). Arrhythmogenic right ventricular cardiomyopathy. Lancet. 373: 1289–1300.
- Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M (1996). Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis?. Circulation. 94: 983–991.
- Bauce B, Basso C, Rampazzo A, Beffagna G, Daliento L, Frigo G, Malacrida S, Settimo L, Danieli G, Thiene G, Nava A (2005). Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J. 26: 1666–1675.
- Beffagna G, De Bortoli M, Nava A, Salamon M, Lorenzon A, Zaccolo M, Mancuso L, Sigalotti L, Bauce B, Occhi G, Basso C, Lanfranchi G, Towbin JA, Thiene G, Danieli GA, Rampazzo A (2007). Missense mutations in desmocollin-2 N-terminus, associated with arrhythmogenic right ventricular cardiomyopathy, affect intracellular localization of desmocollin-2 in vitro. BMC Med Genet. 8: 65.
- Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, Danieli GA, Rampazzo A (2005). Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 65: 366–373.
- Behrens J, Von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W (1996). Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 382: 638–642.
- Ben-Ze'ev A, Geiger B (1998). Differential molecular interactions of beta-catenin and plakoglobin in adhesion, signaling and cancer. Curr Opin Cell Biol. 10: 629–639.
- Bengtsson L, Otto H (2008). LUMA interacts with emerin and influences its distribution at the inner nuclear membrane. J Cell Sci. 121: 536–548.
- Bierkamp C, Schwarz H, Huber O, Kemler R (1999). Desmosomal localization of beta-catenin in the skin of plakoglobin null-mutant mice. Development. 126: 371–381.
- Bolling MC, Jonkman MF (2009). Skin and heart: une liaison dangereuse. Exp Dermatol. 18: 658–668.
- Bolling MC, Veenstra MJ, Jonkman MF, Diercks GF, Curry CJ, Fisher J, Pas HH, Bruckner AL (2010). Lethal acantholytic epidermolysis bullosa due to a novel homozygous deletion in DSP: expanding the phenotype and implications for desmoplakin function in skin and heart. Br J Dermatol. 162: 1388–1394.
- Borrmann CM, Grund C, Kuhn C, Hofmann I, Pieperhoff S, Franke WW (2006). The area composita of adhering junctions connecting heart muscle cells of vertebrates. II. Colocalizations of desmosomal and fascia adhaerens molecules in the intercalated disk.Eur J Cell Biol. 85: 469–485.
- Bowles NE, Ni J, Marcus F, Towbin JA (2002). The detection of cardiotropic viruses in the myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 39: 892–895.
- Brodehl A, Dieding M, Klauke B, Dec E, Madaan S, Huang T, Gargus J, Fatima A, Saric T, Cakar H, Walhorn V, Tonsing K, Skrzipczyk T, Cebulla R, Gerdes D, Schulz U, Gummert J, Svendsen JH, Olesen MS, Anselmetti D, Christensen AH, Kimonis V, Milting H (2013). The novel desmin mutant p.A120D impairs filament formation, prevents intercalated disk localization, and causes sudden cardiac death. Circ Cardiovasc Genet. 6: 615–623.
- Butz S, Stappert J, Weissig H, Kemler R (1992). Plakoglobin and beta-catenin: distinct but closely related. Science. 257: 1142–1144.
- Calabrese F, Basso C, Carturan E, Valente M, Thiene G (2006). Arrhythmogenic right ventricular cardiomyopathy/dysplasia: is there a role for viruses?Cardiovasc Pathol. 15: 11–17.
- Campian ME, Tan HL, van Moerkerken AF, Tukkie R, van Eck-Smit BL, Verberne HJ (2011). Imaging of programmed cell death in arrhythmogenic right ventricle cardiomyopathy/dysplasia. Eur J Nucl Med Mol Imaging. 38: 1500–1506.
- Campian ME, Verberne HJ, Hardziyenka M, de Groot EA, van Moerkerken AF, van Eck-Smit BL, Tan HL (2010). Assessment of inflammation in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur J Nucl Med Mol Imaging. 37: 2079–2085.
- Campuzano S, Balcells L, Villares R, Carramolino L, Garcia-Alonso L, Modolell J (1986). Excess function hairy-wing mutations caused by gypsy and copia insertions within structural genes of the achaete-scute locus of Drosophila. Cell. 44: 303–312.
- Carvajal-Huerta L (1998). Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J Am Acad Dermatol. 39: 418–421.
- Caspi O, Huber I, Gepstein A, Arbel G, Maizels L, Boulos M, Gepstein L (2013). Modeling of arrhythmogenic right ventricular cardiomyopathy with human induced pluripotent stem cells. Circ Cardiovasc Genet. 6: 557–568.
- Cerrone M, Lin X, Zhang M, Agullo-Pascual E, Pfenniger A, Chkourko-Gusky H, Novelli V, Kim C, Tirasawadichai T, Judge DP, Rothenberg E, Chen HS, Napolitano C, Priori S, Delmar M (2013). Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation. 129: 1092–1103.
- Cerrone M, Noorman M, Lin X, Chkourko H, Liang FX, van der Nagel R, Hund T, Birchmeier W, Mohler P, van Veen TA, van Rijen HV, Delmar M (2012). Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res. 95: 460–468.
- Chalabreysse L, Senni F, Bruyere P, Aime B, Ollagnier C, Bozio A, Bouvagnet P (2011). A new hypo/oligodontia syndrome: Carvajal/Naxos syndrome secondary to desmoplakin-dominant mutations. J Dent Res. 90: 58–64.
- Chen J, Nekrasova OE, Patel DM, Klessner JL, Godsel LM, Koetsier JL, Amargo EV, Desai BV, Green KJ (2012). The C-terminal unique region of desmoglein 2 inhibits its internalization via tail–tail interactions. J Cell Biol. 199: 699–711.
- Chen SN, Gurha P, Lombardi R, Ruggiero A, Willerson JT, Marian AJ (2014). The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ Res. 114: 454–468.
- Chen X, Bonne S, Hatzfeld M, van Roy F, Green KJ (2002). Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta -catenin signaling. J Biol Chem. 277: 10512–10522.
- Choi HJ, Weis WI (2011). Crystal structure of a rigid four-spectrin-repeat fragment of the human desmoplakin plakin domain. J Mol Biol. 409: 800–812.
- Christensen AH, Andersen CB, Tybjaerg-Hansen A, Haunso S, Svendsen JH (2011). Mutation analysis and evaluation of the cardiac localization of TMEM43 in arrhythmogenic right ventricular cardiomyopathy. Clin Genet. 80: 256–264.
- Christensen AH, Benn M, Tybjaerg-Hansen A, Haunso S, Svendsen JH (2010). Missense variants in plakophilin-2 in arrhythmogenic right ventricular cardiomyopathy patients—disease-causing or innocent bystanders?Cardiology. 115: 148–154.
- Clemen CS, Herrmann H, Strelkov SV, Schroder R (2013). Desminopathies: pathology and mechanisms. Acta Neuropathol. 125: 47–75.
- Conover GM, Gregorio CC (2011). The desmin coil 1B mutation K190A impairs nebulin Z-disc assembly and destabilizes actin thin filaments. J Cell Sci. 124: 3464–3476.
- Corrado D, Basso C, Pavei A, Michieli P, Schiavon M, Thiene G (2006a). Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 296: 1593–1601.
- Corrado D, Basso C, Pilichou K, Thiene G (2011). Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart. 97: 530–539.
- Corrado D, Basso C, Schiavon M, Pelliccia A, Thiene G (2008). Pre-participation screening of young competitive athletes for prevention of sudden cardiac death. J Am Coll Cardiol. 52: 1981–1989.
- Corrado D, Basso C, Schiavon M, Thiene G (2006b). Does sports activity enhance the risk of sudden cardiac death?J Cardiovasc Med (Hagerstown). 7: 228–233.
- Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom-Lundqvist C, Wlodarska EK, Fontaine G, Camerini F (1997). Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 30: 1512–1520.
- Cowin P, Kapprell HP, Franke WW, Tamkun J, Hynes RO (1986). Plakoglobin: a protein common to different kinds of intercellular adhering junctions. Cell. 46: 1063–1073.
- Cox MG, van der Zwaag PA, van der Werf C, van der Smagt JJ, Noorman M, Bhuiyan ZA, Wiesfeld AC, Volders PG, van Langen IM, Atsma DE, Dooijes D, van den Wijngaard A, Houweling AC, Jongbloed JD, Jordaens L, Cramer MJ, Doevendans PA, de Bakker JM, Wilde AA, van Tintelen JP, Hauer RN (2011). Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation. 123: 2690–2700.
- D’Amati G, Di Gioia CR, Giordano C, Gallo P (2000). Myocyte transdifferentiation: a possible pathogenetic mechanism for arrhythmogenic right ventricular cardiomyopathy. Arch Pathol Lab Med. 124: 287–290.
- Dagvadorj A, Goudeau B, Hilton-Jones D, Blancato JK, Shatunov A, Simon-Casteras M, Squier W, Nagle JW, Goldfarb LG, Vicart P (2003). Respiratory insufficiency in desminopathy patients caused by introduction of proline residues in desmin c-terminal alpha-helical segment. Muscle Nerve. 27: 669–675.
- Dalakas MC, Dagvadorj A, Goudeau B, Park KY, Takeda K, Simon-Casteras M, Vasconcelos O, Sambuughin N, Shatunov A, Nagle JW, Sivakumar K, Vicart P, Goldfarb LG (2003). Progressive skeletal myopathy, a phenotypic variant of desmin myopathy associated with desmin mutations. Neuromuscul Disord. 13: 252–258.
- Delmar M, McKenna WJ (2010). The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 107: 700–714.
- Delva E, Tucker DK, Kowalczyk AP (2009). The desmosome. Cold Spring Harb Perspect Biol. 1: a002543.
- Desplantez T, Mccain ML, Beauchamp P, Rigoli G, Rothen-Rutishauser B, Parker KK, Kleber AG (2012). Connexin43 ablation in foetal atrial myocytes decreases electrical coupling, partner connexins, and sodium current. Cardiovasc Res. 94: 58–65.
- Elliott P (2008). The 2006 American Heart Association classification of cardiomyopathies is not the gold standard. Circ Heart Fail. 1: 77–79; discussion 80.
- Elliott P, O'mahony C, Syrris P, Evans A, Rivera Sorensen C, Sheppard MN, Carr-White G, Pantazis A, McKenna WJ (2010). Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ Cardiovasc Genet. 3: 314–322.
- Eshkind L, Tian Q, Schmidt A, Franke WW, Windoffer R, Leube RE (2002). Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur J Cell Biol. 81: 592–598.
- Fidler LM, Wilson GJ, Liu F, Cui X, Scherer SW, Taylor GP, Hamilton RM (2009). Abnormal connexin43 in arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 mutations. J Cell Mol Med. 13: 4219–4228.
- Fornes P, Ratel S, Lecomte D (1998). Pathology of arrhythmogenic right ventricular cardiomyopathy/dysplasia—an autopsy study of 20 forensic cases. J Forensic Sci. 43: 777–783.
- Frank R, Fontaine G, Vedel J, Mialet G, Sol C, Guiraudon G, Grosgogeat Y (1978). Electrocardiology of 4 cases of right ventricular dysplasia inducing arrhythmia. Arch Mal Coeur Vaiss. 71: 963–972.
- Franke WW, Borrmann CM, Grund C, Pieperhoff S (2006). The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins.Eur J Cell Biol. 85: 69–82.
- Franke WW, Schumacher H, Borrmann CM, Grund C, Winter-Simanowski S, Schlechter T, Pieperhoff S, Hofmann I (2007). The area composita of adhering junctions connecting heart muscle cells of vertebrates—III: assembly and disintegration of intercalated disks in rat cardiomyocytes growing in culture. Eur J Cell Biol. 86: 127–142.
- Fujita S, Terasaki F, Otsuka K, Katashima T, Kanzaki Y, Kawamura K, Tanaka T, Kitaura Y (2008). Markedly increased intracellular lipid droplets and disruption of intercellular junctions in biopsied myocardium from a patient with arrhythmogenic right ventricular cardiomyopathy. Heart Vessels. 23: 440–444.
- Gaertner A, Klauke B, Stork I, Niehaus K, Niemann G, Gummert J, Milting H (2012). In vitro functional analyses of arrhythmogenic right ventricular cardiomyopathy-associated desmoglein-2-missense variations. PLoS One. 7: e47097.
- Gallicano GI, Bauer C, Fuchs E (2001). Rescuing desmoplakin function in extra-embryonic ectoderm reveals the importance of this protein in embryonic heart, neuroepithelium, skin and vasculature. Development. 128: 929–941.
- Gallicano GI, Kouklis P, Bauer C, Yin M, Vasioukhin V, Degenstein L, Fuchs E (1998). Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J Cell Biol. 143: 2009–2022.
- Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ (2006). Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 116: 2012–2021.
- Garcia-Pavia P, Syrris P, Salas C, Evans A, Mirelis JG, Cobo-Marcos M, Vilches C, Bornstein B, Segovia J, Alonso-Pulpon L, Elliott PM (2011). Desmosomal protein gene mutations in patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation: a clinicopathological study. Heart. 97: 1744–1752.
- Gard JJ, Yamada K, Green KG, Eloff BC, Rosenbaum DS, Wang X, Robbins J, Schuessler RB, Yamada KA, Saffitz JE (2005). Remodeling of gap junctions and slow conduction in a mouse model of desmin-related cardiomyopathy. Cardiovasc Res. 67: 539–547.
- Gattenlohner S, Demmer P, Oberhoff M, Ertl G (2009). Acute episode of an arrhythmogenic right ventricular cardiomyopathy with vast necroses exclusively in right ventricular myocardium. Eur Heart J. 30: 2430.
- Gehmlich K, Asimaki A, Cahill TJ, Ehler E, Syrris P, Zachara E, Re F, Avella A, Monserrat L, Saffitz JE, McKenna WJ (2010). Novel missense mutations in exon 15 of desmoglein-2: role of the intracellular cadherin segment in arrhythmogenic right ventricular cardiomyopathy?Heart Rhythm. 7: 1446–1453.
- Gehmlich K, Lambiase PD, Asimaki A, Ciaccio EJ, Ehler E, Syrris P, Saffitz JE, McKenna WJ (2011a). A novel desmocollin-2 mutation reveals insights into the molecular link between desmosomes and gap junctions. Heart Rhythm. 8: 711–718.
- Gehmlich K, Syrris P, Peskett E, Evans A, Ehler E, Asimaki A, Anastasakis A, Tsatsopoulou A, Vouliotis AI, Stefanadis C, Saffitz JE, Protonotarios N, McKenna WJ (2011b). Mechanistic insights into arrhythmogenic right ventricular cardiomyopathy caused by desmocollin-2 mutations. Cardiovasc Res. 90: 77–87.
- Getsios S, Waschke J, Borradori L, Hertl M, Muller EJ (2010). From cell signaling to novel therapeutic concepts: international pemphigus meeting on advances in pemphigus research and therapy. J Invest Dermatol. 130: 1764–1768.
- Gittenberger-de Groot AC, Winter EM, Poelmann RE (2010). Epicardium-derived cells (EPDCs) in development, cardiac disease and repair of ischemia. J Cell Mol Med. 14: 1056–1060.
- Godsel LM, Dubash AD, Bass-Zubek AE, Amargo EV, Klessner JL, Hobbs RP, Chen X, Green KJ (2010). Plakophilin 2 couples actomyosin remodeling to desmosomal plaque assembly via RhoA. Mol Biol Cell. 21: 2844–2859.
- Goldfarb LG, Park KY, Cervenakova L, Gorokhova S, Lee HS, Vasconcelos O, Nagle JW, Semino-Mora C, Sivakumar K, Dalakas MC (1998). Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. 19: 402–403.
- Goldfarb LG, Vicart P, Goebel HH, Dalakas MC (2004). Desmin myopathy. Brain. 127: 723–734.
- Gomes J, Finlay M, Ahmed AK, Ciaccio EJ, Asimaki A, Saffitz JE, Quarta G, Nobles M, Syrris P, Chaubey S, McKenna WJ, Tinker A, Lambiase PD (2012). Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur Heart J. 33: 1942–1953.
- Goossens S, Janssens B, Bonne S, de Rycke R, Braet F, van Hengel J, van Roy F (2007). A unique and specific interaction between alphaT-catenin and plakophilin-2 in the area composita, the mixed-type junctional structure of cardiac intercalated discs. J Cell Sci. 120: 2126–2136.
- Goudeau B, Dagvadorj A, Rodrigues-Lima F, Nedellec P, Casteras-Simon M, Perret E, Langlois S, Goldfarb L, Vicart P (2001). Structural and functional analysis of a new desmin variant causing desmin-related myopathy. Hum Mutat. 18: 388–396.
- Groeneweg JA, van der Zwaag PA, Jongbloed JD, Cox MG, Vreeker A, de Boer RA, van der Heijden JF, van Veen TA, McKenna WJ, van Tintelen JP, Dooijes D, Hauer RN (2013). Left-dominant arrhythmogenic cardiomyopathy in a large family: associated desmosomal or nondesmosomal genotype?Heart Rhythm. 10: 548–559.
- Grossmann KS, Grund C, Huelsken J, Behrend M, Erdmann B, Franke WW, Birchmeier W (2004). Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol. 167: 149–160.
- Grumbach IM, Heim A, Vonhof S, Stille-Siegener M, Mall G, Gonska BD, Kreuzer H, Andreas S, Figulla HR (1998). Coxsackievirus genome in myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Cardiology. 89: 241–245.
- H Fischer A, van der Loo B, Shar MG, Zbinden R, Duru F, Brunckhorst C, Rousson V, Delacretaz YE, Stuber T, Oechslin EN, Follath F, Jenni R (2008). Serological evidence for the association of Bartonella henselae infection with arrhythmogenic right ventricular cardiomyopathy. Clin Cardiol. 31: 469–471.
- Harmon RM, Green KJ (2013). Structural and functional diversity of desmosomes. Cell Commun Adhes. 20: 171–187.
- Harvey PA, Leinwand LA (2011). The cell biology of disease: cellular mechanisms of cardiomyopathy. J Cell Biol. 194: 355–365.
- Hatzfeld M (2007). Plakophilins: Multifunctional proteins or just regulators of desmosomal adhesion?Biochim Biophys Acta. 1773: 69–77.
- Haywood AF, Merner ND, Hodgkinson KA, Houston J, Syrris P, Booth V, Connors S, Pantazis A, Quarta G, Elliott P, McKenna W, Young TL (2013). Recurrent missense mutations in TMEM43 (ARVD5) due to founder effects cause arrhythmogenic cardiomyopathies in the UK and Canada. Eur Heart J. 34: 1002–1011.
- Hedberg C, Melberg A, Kuhl A, Jenne D, Oldfors A (2012). Autosomal dominant myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy 7 is caused by a DES mutation. Eur J Hum Genet. 20: 984–985.
- Herren T, Gerber PA, Duru F (2009). Arrhythmogenic right ventricular cardiomyopathy/dysplasia: a not so rare “disease of the desmosome” with multiple clinical presentations. Clin Res Cardiol. 98: 141–158.
- Hobbs RP, Amargo EV, Somasundaram A, Simpson CL, Prakriya M, Denning MF, Green KJ (2011). The calcium ATPase SERCA2 regulates desmoplakin dynamics and intercellular adhesive strength through modulation of PKC & alpha; signaling. FASEB J. 25: 990–1001.
- Hobbs RP, Han SY, van der Zwaag PA, Bolling MC, Jongbloed JD, Jonkman MF, Getsios S, Paller AS, Green KJ (2010). Insights from a desmoplakin mutation identified in lethal acantholytic epidermolysis bullosa. J Invest Dermatol. 130: 2680–2683.
- Hofmann I, Casella M, Schnolzer M, Schlechter T, Spring H, Franke WW (2006). Identification of the junctional plaque protein plakophilin 3 in cytoplasmic particles containing RNA-binding proteins and the recruitment of plakophilins 1 and 3 to stress granules. Mol Biol Cell. 17: 1388–1398.
- Hollrigl A, Hofner M, Stary M, Weitzer G (2007). Differentiation of cardiomyocytes requires functional serine residues within the amino-terminal domain of desmin. Differentiation. 75: 616–626.
- Hu P, Berkowitz P, O'keefe EJ, Rubenstein DS (2003). Keratinocyte adherens junctions initiate nuclear signaling by translocation of plakoglobin from the membrane to the nucleus. J Invest Dermatol. 121: 242–251.
- Huang H, Asimaki A, Lo D, McKenna W, Saffitz J (2008). Disparate effects of different mutations in plakoglobin on cell mechanical behavior. Cell Motil Cytoskeleton. 65: 964–978.
- Huber O, Korn R, Mclaughlin J, Ohsugi M, Herrmann BG, Kemler R (1996). Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev. 59: 3–10.
- Jansen JA, Noorman M, Musa H, Stein M, de Jong S, van der Nagel R, Hund TJ, Mohler PJ, Vos MA, van Veen TA, de Bakker JM, Delmar M, van Rijen HV (2012). Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm. 9: 600–607.
- Jonkman MF, Pasmooij AM, Pasmans SG, van den Berg MP, Ter Horst HJ, Timmer A, Pas HH (2005). Loss of desmoplakin tail causes lethal acantholytic epidermolysis bullosa. Am J Hum Genet. 77: 653–660.
- Joshi-Mukherjee R, Coombs W, Musa H, Oxford E, Taffet S, Delmar M (2008). Characterization of the molecular phenotype of two arrhythmogenic right ventricular cardiomyopathy (ARVC)-related plakophilin-2 (PKP2) mutations. Heart Rhythm. 5: 1715–1723.
- Kaplan SR, Gard JJ, Carvajal-Huerta L, Ruiz-Cabezas JC, Thiene G, Saffitz JE (2004a). Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc Pathol. 13: 26–32.
- Kaplan SR, Gard JJ, Protonotarios N, Tsatsopoulou A, Spiliopoulou C, Anastasakis A, Squarcioni CP, McKenna WJ, Thiene G, Basso C, Brousse N, Fontaine G, Saffitz JE (2004b). Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm. 1: 3–11.
- Kapoun AM, Liang F, O'young G, Damm DL, Quon D, White RT, Munson K, Lam A, Schreiner GF, Protter AA (2004). B-type natriuretic peptide exerts broad functional opposition to transforming growth factor-beta in primary human cardiac fibroblasts: fibrosis, myofibroblast conversion, proliferation, and inflammation. Circ Res. 94: 453–461.
- Kapplinger JD, Landstrom AP, Salisbury BA, Callis TE, Pollevick GD, Tester DJ, Cox MG, Bhuiyan Z, Bikker H, Wiesfeld AC, Hauer RN, van Tintelen JP, Jongbloed JD, Calkins H, Judge DP, Wilde AA, Ackerman MJ (2011). Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol. 57: 2317–2327.
- Kim C, Wong J, Wen J, Wang S, Wang C, Spiering S, Kan NG, Forcales S, Puri PL, Leone TC, Marine JE, Calkins H, Kelly DP, Judge DP, Chen HS (2013). Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 494: 105–110.
- Kirchhof P, Breithardt G, Eckardt L (2006). Primary prevention of sudden cardiac death. Heart. 92: 1873–1878.
- Kirchner F, Schuetz A, Boldt LH, Martens K, Dittmar G, Haverkamp W, Thierfelder L, Heinemann U, Gerull B (2012). Molecular insights into arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 missense mutations. Circ Cardiovasc Genet. 5: 400–411.
- Klauke B, Kossmann S, Gaertner A, Brand K, Stork I, Brodehl A, Dieding M, Walhorn V, Anselmetti D, Gerdes D, Bohms B, Schulz U, Zu Knyphausen E, Vorgerd M, Gummert J, Milting H (2010). De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet. 19: 4595–4607.
- Koetsier JL, Amargo EV, Todorovic V, Green KJ, Godsel LM (2014). Plakophilin 2 affects cell migration by modulating focal adhesion dynamics and integrin protein expression. J Inves Dermatol. 134: 112–122.
- Kowalczyk AP, Navarro P, Dejana E, Bornslaeger EA, Green KJ, Kopp DS, Borgwardt JE (1998). VE-cadherin and desmoplakin are assembled into dermal microvascular endothelial intercellular junctions: a pivotal role for plakoglobin in the recruitment of desmoplakin to intercellular junctions. J Cell Sci. 111(Pt 20): 3045–3057.
- Krusche CA, Holthofer B, Hofe V, van de Sandt AM, Eshkind L, Bockamp E, Merx MW, Kant S, Windoffer R, Leube RE (2011). Desmoglein 2 mutant mice develop cardiac fibrosis and dilation. Basic Res Cardiol. 106: 617–633.
- Lancisi GM (1736). De motu cordis et aneurysmatibus. Naples.
- Lapouge K, Fontao L, Champliaud MF, Jaunin F, Frias MA, Favre B, Paulin D, Green KJ, Borradori L (2006). New insights into the molecular basis of desmoplakin- and desmin-related cardiomyopathies. J Cell Sci. 119: 4974–4985.
- Larsen MK, Nissen PH, Berge KE, Leren TP, Kristensen IB, Jensen HK, Banner J (2012). Molecular autopsy in young sudden cardiac death victims with suspected cardiomyopathy. Forensic Sci Int. 219: 33–38.
- Leask A, Abraham DJ (2004). TGF-beta signaling and the fibrotic response. FASEB J. 18: 816–827.
- Li D, Liu Y, Maruyama M, Zhu W, Chen H, Zhang W, Reuter S, Lin SF, Haneline LS, Field LJ, Chen PS, Shou W (2011a). Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Hum Mol Genet. 20: 4582–4596.
- Li D, Tapscoft T, Gonzalez O, Burch PE, Quinones MA, Zoghbi WA, Hill R, Bachinski LL, Mann DL, Roberts R (1999). Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation. 100: 461–464.
- Li D, Zhang W, Liu Y, Haneline LS, Shou W (2012a). Lack of plakoglobin in epidermis leads to keratoderma. J Biol Chem. 287: 10435–10443.
- Li J, Goossens S, van Hengel J, Gao E, Cheng L, Tyberghein K, Shang X, De Rycke R, van Roy F, Radice GL (2012b). Loss of alphaT-catenin alters the hybrid adhering junctions in the heart and leads to dilated cardiomyopathy and ventricular arrhythmia following acute ischemia. J Cell Sci. 125: 1058–1067.
- Li J, Swope D, Raess N, Cheng L, Muller EJ, Radice GL (2011b). Cardiac tissue-restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of {beta}-catenin signaling. Mol Cell Biol. 31: 1134–1144.
- Li Mura IE, Bauce B, Nava A, Fanciulli M, Vazza G, Mazzotti E, Rigato I, De Bortoli M, Beffagna G, Lorenzon A, Calore M, Dazzo E, Nobile C, Mostacciuolo ML, Corrado D, Basso C, Daliento L, Thiene G, Rampazzo A (2013). Identification of a PKP2 gene deletion in a family with arrhythmogenic right ventricular cardiomyopathy. Eur J Hum Genet. 21: 1226–1231.
- Li MW, Mruk DD, Lee WM, Cheng CY (2009). Connexin 43 and plakophilin-2 as a protein complex that regulates blood-testis barrier dynamics. Proc Natl Acad Sci U S A. 106: 10213–10218.
- Li Z, Colucci-Guyon E, Pincon-Raymond M, Mericskay M, Pournin S, Paulin D, Babinet C (1996). Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev Biol. 175: 362–366.
- Lombardi R, Da Graca Cabreira-Hansen M, Bell A, Fromm RR, Willerson JT, Marian AJ (2011). Nuclear plakoglobin is essential for differentiation of cardiac progenitor cells to adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ Res. 109: 1342–1353.
- Lombardi R, Dong J, Rodriguez G, Bell A, Leung TK, Schwartz RJ, Willerson JT, Brugada R, Marian AJ (2009). Genetic fate mapping identifies second heart field progenitor cells as a source of adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circ Res. 104: 1076–1084.
- Lorenzon A, Beffagna G, Bauce B, De Bortoli M, Li Mura IE, Calore M, Dazzo E, Basso C, Nava A, Thiene G, Rampazzo A (2013). Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 111: 400–405.
- Lyon RC, Mezzano V, Wright AT, Pfeiffer E, Chuang J, Banares K, Castaneda A, Ouyang K, Cui L, Contu R, Gu Y, Evans SM, Omens JH, Peterson KL, Mcculloch AD, Sheikh F (2013). Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum Mol Genet. 23: 1134–1150.
- Ma D, Wei H, Lu J, Ho S, Zhang G, Sun X, Oh Y, Tan SH, Ng ML, Shim W, Wong P, Liew R (2013). Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 34: 1122–1133.
- Mahoney MG, Sadowski S, Brennan D, Pikander P, Saukko P, Wahl J, Aho H, Heikinheimo K, Bruckner-Tuderman L, Fertala A, Peltonen J, Uitto J, Peltonen S (2010). Compound heterozygous desmoplakin mutations result in a phenotype with a combination of myocardial, skin, hair, and enamel abnormalities. J Invest Dermatol. 130: 968–978.
- Mallat Z, Tedgui A, Fontaliran F, Frank R, Durigon M, Fontaine G (1996). Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N Engl J Med. 335: 1190–1196.
- Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y (1982). Right ventricular dysplasia: a report of 24 adult cases. Circulation. 65: 384–398.
- Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W (2010). Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 121: 1533–1541.
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB, American Heart Association, Council on Clinical Cardiology, Heart Failure and Transplantation Committee, Quality of Care and Outcomes Research and Functional Genomicsand Translational Biology Interdisciplinary Working Groups, Council on Epidemiologyand Prevention (2006). Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 113: 1807–1816.
- Matthes SA, Taffet S, Delmar M (2011). Plakophilin-2 and the migration, differentiation and transformation of cells derived from the epicardium of neonatal rat hearts. Cell Commun Adhes. 18: 73–84.
- Mavroidis M, Panagopoulou P, Kostavasili I, Weisleder N, Capetanaki Y (2008). A missense mutation in desmin tail domain linked to human dilated cardiomyopathy promotes cleavage of the head domain and abolishes its Z-disc localization. FASEB J. 22: 3318–3327.
- McCrea PD, Turck CW, Gumbiner B (1991). A homolog of the armadillo protein in Drosophila (plakoglobin) associated with E-cadherin. Science. 254: 1359–1361.
- McGrath JA (1999). Hereditary diseases of desmosomes. J Dermatol Sci. 20: 85–91.
- McGrath JA, Wessagowit V (2005). Human hair abnormalities resulting from inherited desmosome gene mutations. Keio J Med. 54: 72–79.
- McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, Camerini F (1994). Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology.Br Heart J. 71: 215–218.
- McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ (2000). Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. 355: 2119–2124.
- McLendon PM, Robbins J (2011). Desmin-related cardiomyopathy: an unfolding story. Am J Physiol Heart Circ Physiol. 301: H1220–H1228.
- Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, Gallagher B, Morris-Larkin L, Bassett AS, Parfrey PS, Young TL (2008). Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 82: 809–821.
- Mertens C, Hofmann I, Wang Z, Teichmann M, Sepehri Chong S, Schnolzer M, Franke WW (2001). Nuclear particles containing RNA polymerase III complexes associated with the junctional plaque protein plakophilin 2. Proc Natl Acad Sci U S A. 98: 7795–7800.
- Miller RK, Hong JY, Munoz WA, Mccrea PD (2013). Beta-catenin versus the other armadillo catenins: assessing our current view of canonical Wnt signaling. Prog Mol Biol Transl Sci. 116: 387–407.
- Milner DJ, Weitzer G, Tran D, Bradley A, Capetanaki Y (1996). Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol. 134: 1255–1270.
- Miravet S, Piedra J, Miro F, Itarte E, Garcia de Herreros A, Dunach M (2002). The transcriptional factor Tcf-4 contains different binding sites for beta-catenin and plakoglobin. J Biol Chem. 277: 1884–1891.
- Muller J, Ritt DA, Copeland TD, Morrison DK (2003). Functional analysis of C-TAK1 substrate binding and identification of PKP2 as a new C-TAK1 substrate. EMBO J. 22: 4431–4442.
- Munoz WA, Lee M, Miller RK, Ahmed Z, Ji H, Link TM, Lee GR, Kloc M, Ladbury JE, Mccrea PD (2014). Plakophilin-3 catenin associates with the ETV1/ER81 transcription factor to positively modulate gene activity. PLoS One. 9: e86784.
- Munoz-Marmol AM, Strasser G, Isamat M, Coulombe PA, Yang Y, Roca X, Vela E, Mate JL, Coll J, Fernandez-Figueras MT, Navas-Palacios JJ, Ariza A, Fuchs E (1998). A dysfunctional desmin mutation in a patient with severe generalized myopathy. Proc Natl Acad Sci U S A. 95: 11312–11317.
- Nagata M, Hiroe M, Ishiyama S, Nishikawa T, Sakomura Y, Kasanuki H, Toyosaki T, Marumo F (2000). Apoptotic cell death in arrhythmogenic right ventricular cardiomyopathy: a comparative study with idiopathic sustained ventricular tachycardia. Jpn Heart J. 41: 733–741.
- Nishikawa T, Ishiyama S, Nagata M, Sakomura Y, Nakazawa M, Momma K, Hiroe M, Kasajima T (1999). Programmed cell death in the myocardium of arrhythmogenic right ventricular cardiomyopathy in children and adults. Cardiovasc Pathol. 8: 185–189.
- Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, Whittock N, Leigh IM, Stevens HP, Kelsell DP (2000). Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 9: 2761–2766.
- Norman M, Simpson M, Mogensen J, Shaw A, Hughes S, Syrris P, Sen-Chowdhry S, Rowland E, Crosby A, McKenna WJ (2005). Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation. 112: 636–642.
- Okabe M, Fukuda K, Nakashima Y, Arakawa K, Kikuchi M (1995). An isolated left ventricular lesion associated with left ventricular tachycardia: arrhythmogenic “left” ventricular dysplasia?Jpn Circ J. 59: 49–54.
- Omary MB (2009). “IF-pathies”: a broad spectrum of intermediate filament-associated diseases. J Clin Invest. 119: 1756–1762.
- Otten E, Asimaki A, Maass A, van Langen IM, van der Wal A, de Jonge N, van den Berg MP, Saffitz JE, Wilde AA, Jongbloed JD, van Tintelen JP (2010). Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm. 7: 1058–1064.
- Oxford EM, Musa H, Maass K, Coombs W, Taffet SM, Delmar M (2007). Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ Res. 101: 703–711.
- Palka HL, Green KJ (1997). Roles of plakoglobin end domains in desmosome assembly. J Cell Sci. 110 (Pt 19): 2359–2371.
- Pica EC, Kathirvel P, Pramono ZA, Lai PS, Yee WC (2008). Characterization of a novel S13F desmin mutation associated with desmin myopathy and heart block in a Chinese family. Neuromuscul Disord. 18: 178–182.
- Pieperhoff S, Borrmann C, Grund C, Barth M, Rizzo S, Franke WW (2010). The area composita of adhering junctions connecting heart muscle cells of vertebrates. VII. The different types of lateral junctions between the special cardiomyocytes of the conduction system of ovine and bovine hearts.Eur J Cell Biol. 89: 365–378.
- Pieperhoff S, Franke WW (2007). The area composita of adhering junctions connecting heart muscle cells of vertebrates—IV: coalescence and amalgamation of desmosomal and adhaerens junction components—late processes in mammalian heart development. Eur J Cell Biol. 86: 377–391.
- Pieperhoff S, Franke WW (2008). The area composita of adhering junctions connecting heart muscle cells of vertebrates. VI. Different precursor structures in non-mammalian species. Eur J Cell Biol. 87: 413–430.
- Pieperhoff S, Schumacher H, Franke WW (2008). The area composita of adhering junctions connecting heart muscle cells of vertebrates. V. The importance of plakophilin-2 demonstrated by small interference RNA-mediated knockdown in cultured rat cardiomyocytes.Eur J Cell Biol. 87: 399–411.
- Pilichou K, Remme CA, Basso C, Campian ME, Rizzo S, Barnett P, Scicluna BP, Bauce B, van den Hoff MJ, de Bakker JM, Tan HL, Valente M, Nava A, Wilde AA, Moorman AF, Thiene G, Bezzina CR (2009). Myocyte necrosis underlies progressive myocardial dystrophy in mouse Dsg2-related arrhythmogenic right ventricular cardiomyopathy. J Exp Med. 206: 1787–1802.
- Protonotarios N, Tsatsopoulou A, Patsourakos P, Alexopoulos D, Gezerlis P, Simitsis S, Scampardonis G (1986). Cardiac abnormalities in familial palmoplantar keratosis. Br Heart J. 56: 321–326.
- Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM, Lakdawala NK, Mcdermott G, White ET, Rehm HL, Lebo M, Funke BH (2014). The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. (Epub ahead of print).
- Quarta G, Syrris P, Ashworth M, Jenkins S, Zuborne Alapi K, Morgan J, Muir A, Pantazis A, McKenna WJ, Elliott PM (2012). Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 33: 1128–1136.
- Rao BH, Reddy IS, Chandra KS (1996). Familial occurrence of a rare combination of dilated cardiomyopathy with palmoplantar keratoderma and curly hair. Indian Heart J. 48: 161–162.
- Rasmussen TB, Hansen J, Nissen PH, Palmfeldt J, Dalager S, Jensen UB, Kim WY, Heickendorff L, Molgaard H, Jensen HK, Sorensen KE, Baandrup UT, Bross P, Mogensen J (2013a). Protein expression studies of desmoplakin mutations in cardiomyopathy patients reveal different molecular disease mechanisms. Clin Genet. 84: 20–30.
- Rasmussen TB, Palmfeldt J, Nissen PH, Magnoni R, Dalager S, Jensen UB, Kim WY, Heickendorff L, Molgaard H, Jensen HK, Baandrup UT, Bross P, Mogensen J (2013b). Mutated desmoglein-2 proteins are incorporated into desmosomes and exhibit dominant-negative effects in arrhythmogenic right ventricular cardiomyopathy. Hum Mutat. 34: 697–705.
- Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P (1996). Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation. 93: 841–842.
- Rickelt S, Pieperhoff S (2012). Mutations with pathogenic potential in proteins located in or at the composite junctions of the intercalated disk connecting mammalian cardiomyocytes: a reference thesaurus for arrhythmogenic cardiomyopathies and for Naxos and Carvajal diseases. Cell Tissue Res. 348: 325–333.
- Rickelt S, Rizzo S, Doerflinger Y, Zentgraf H, Basso C, Gerosa G, Thiene G, Moll R, Franke WW (2010). A novel kind of tumor type-characteristic junction: plakophilin-2 as a major protein of adherens junctions in cardiac myxomata. Mod Pathol. 23: 1429–1437.
- Rigato I, Bauce B, Rampazzo A, Zorzi A, Pilichou K, Mazzotti E, Migliore F, Marra MP, Lorenzon A, De Bortoli M, Calore M, Nava A, Daliento L, Gregori D, Iliceto S, Thiene G, Basso C, Corrado D (2013). Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 6: 533–542.
- Rizzo S, Lodder EM, Verkerk AO, Wolswinkel R, Beekman L, Pilichou K, Basso C, Remme CA, Thiene G, Bezzina CR (2012). Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res. 95: 409–418.
- Ruiz P, Brinkmann V, Ledermann B, Behrend M, Grund C, Thalhammer C, Vogel F, Birchmeier C, Gunthert U, Franke WW, Birchmeier W (1996). Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol. 135: 215–225.
- Sato PY, Coombs W, Lin X, Nekrasova O, Green KJ, Isom LL, Taffet SM, Delmar M (2011). Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ Res. 109: 193–201.
- Sato PY, Musa H, Coombs W, Guerrero-Serna G, Patino GA, Taffet SM, Isom LL, Delmar M (2009). Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 105: 523–526.
- Schmidt A, Koch PJ (2007). Desmosomes: just cell adhesion or is there more?Cell Adh Migr. 1: 28–32.
- Schmidt A, Langbein L, Rode M, Pratzel S, Zimbelmann R, Franke WW (1997). Plakophilins 1a and 1b: widespread nuclear proteins recruited in specific epithelial cells as desmosomal plaque components. Cell Tissue Res. 290: 481–499.
- Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ (2008). Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 52: 2175–2187.
- Silvano M, Corrado D, Kobe J, Monnig G, Basso C, Thiene G, Eckardt L (2013). Risk stratification in arrhythmogenic right ventricular cardiomyopathy. Herzschrittmacherther Elektrophysiol. 24: 202–208.
- Simcha I, Shtutman M, Salomon D, Zhurinsky J, Sadot E, Geiger B, Ben-Ze'ev A (1998). Differential nuclear translocation and transactivation potential of beta-catenin and plakoglobin. J Cell Biol. 141: 1433–1448.
- Simpson CL, Patel DM, Green KJ (2011). Deconstructing the skin: cytoarchitectural determinants of epidermal morphogenesis. Nat Rev Mol Cell Biol. 12: 565–580.
- Sjoberg G, Saavedra-Matiz CA, Rosen DR, Wijsman EM, Borg K, Horowitz SH, Sejersen T (1999). A missense mutation in the desmin rod domain is associated with autosomal dominant distal myopathy, and exerts a dominant negative effect on filament formation. Hum Mol Genet. 8: 2191–2198.
- Smith FJ, Wilson NJ, Moss C, Dopping-Hepenstal P, McGrath J (2012). Compound heterozygous mutations in desmoplakin cause skin fragility and woolly hair. Br J Dermatol. 166: 894–896.
- Suzuki H, Sumiyoshi M, Kawai S, Takagi A, Wada A, Nakazato Y, Daida H, Sakurai H, Yamaguchi H (2000). Arrhythmogenic right ventricular cardiomyopathy with an initial manifestation of severe left ventricular impairment and normal contraction of the right ventricle. Jpn Circ J. 64: 209–213.
- Tabib A, Loire R, Chalabreysse L, Meyronnet D, Miras A, Malicier D, Thivolet F, Chevalier P, Bouvagnet P (2003). Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation. 108: 3000–3005.
- Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, Pinamonti B, Salcedo EE, Sauer W, Pyxaras S, Anderson B, Simon B, Bogomolovas J, Labeit S, Granzier H, Mestroni L (2011). Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation. 124: 876–885.
- Te Riele AS, James CA, Philips B, Rastegar N, Bhonsale A, Groeneweg JA, Murray B, Tichnell C, Judge DP, van der Heijden JF, Cramer MJ, Velthuis BK, Bluemke DA, Zimmerman SL, Kamel IR, Hauer RN, Calkins H, Tandri H (2013). Mutation-positive arrhythmogenic right ventricular dysplasia/cardiomyopathy: the triangle of dysplasia displaced. J Cardiovasc Electrophysiol. 24: 1311–1320.
- Todorovic V, Desai BV, Patterson MJ, Amargo EV, Dubash AD, Yin T, Jones JC, Green KJ (2010). Plakoglobin regulates cell motility through Rho- and fibronectin-dependent Src signaling. J Cell Sci. 123: 3576–3586.
- Tse HF, Ho JC, Choi SW, Lee YK, Butler AW, Ng KM, Siu CW, Simpson MA, Lai WH, Chan YC, Au KW, Zhang J, Lay KW, Esteban MA, Nicholls JM, Colman A, Sham PC (2013). Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum Mol Genet. 22: 1395–1403.
- Uhl HS (1952). A previously undescribed congenital malformation of the heart: almost total absence of the myocardium of the right ventricle. Bull Johns Hopkins Hosp. 91: 197–209.
- Valente M, Calabrese F, Thiene G, Angelini A, Basso C, Nava A, Rossi L (1998). In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am J Pathol. 152: 479–484.
- van der Smagt JJ, van der Zwaag PA, van Tintelen JP, Cox MG, Wilde AA, van Langen IM, Ummels A, Hennekam FA, Dooijes D, Gerbens F, Bikker H, Hauer RN, Doevendans PA (2012). Clinical and genetic characterization of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy caused by a plakophilin-2 splice mutation. Cardiology. 123: 181–189.
- van der Zwaag PA, Jongbloed JD, van den Berg MP, van der Smagt JJ, Jongbloed R, Bikker H, Hofstra RM, van Tintelen JP (2009). A genetic variants database for arrhythmogenic right ventricular dysplasia/cardiomyopathy. Hum Mutat. 30: 1278–1283.
- van der Zwaag PA, van Rijsingen IA, Asimaki A, Jongbloed JD, van Veldhuisen DJ, Wiesfeld AC, Cox MG, van Lochem LT, de Boer RA, Hofstra RM, Christiaans I, van Spaendonck-Zwarts KY, Lekanne Dit Deprez RH, Judge DP, Calkins H, Suurmeijer AJ, Hauer RN, Saffitz JE, Wilde AA, van den Berg MP, van Tintelen JP (2012). Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 14: 1199–1207.
- van der Zwaag PA, van Rijsingen IA, de Ruiter R, Nannenberg EA, Groeneweg JA, Post JG, Hauer RN, van Gelder IC, van den Berg MP, van der Harst P, Wilde AA, van Tintelen JP (2013). Recurrent and founder mutations in the Netherlands-Phospholamban p.Arg14del mutation causes arrhythmogenic cardiomyopathy. Neth Heart J. 21: 286–293.
- van Hengel J, Calore M, Bauce B, Dazzo E, Mazzotti E, De Bortoli M, Lorenzon A, Li Mura IE, Beffagna G, Rigato I, Vleeschouwers M, Tyberghein K, Hulpiau P, van Hamme E, Zaglia T, Corrado D, Basso C, Thiene G, Daliento L, Nava A, van Roy F, Rampazzo A (2013). Mutations in the area composita protein alphaT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 34: 201–210.
- van Hengel J, Gohon L, Bruyneel E, Vermeulen S, Cornelissen M, Mareel M, Von Roy F (1997). Protein kinase C activation upregulates intercellular adhesion of alpha-catenin-negative human colon cancer cell variants via induction of desmosomes. J Cell Biol. 137: 1103–1116.
- van Spaendonck-Zwarts KY, van Hessem L, Jongbloed JD, de Walle HE, Capetanaki Y, van der Kooi AJ, van Langen IM, van den Berg MP, van Tintelen JP (2011). Desmin-related myopathy. Clin Genet. 80: 354–366.
- van Tintelen JP, van Gelder IC, Asimaki A, Suurmeijer AJ, Wiesfeld AC, Jongbloed JD, van den Wijngaard A, Kuks JB, van Spaendonck-Zwarts KY, Notermans N, Boven L, van den Heuvel F, Veenstra-Knol HE, Saffitz JE, Hofstra RM, van den Berg MP (2009). Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm. 6: 1574–1583.
- Varga J, Jimenez SA (1986). Stimulation of normal human fibroblast collagen production and processing by transforming growth factor-beta. Biochem Biophys Res Commun. 138: 974–980.
- Vasioukhin V, Bowers E, Bauer C, Degenstein L, Fuchs E (2001). Desmoplakin is essential in epidermal sheet formation. Nat Cell Biol. 3: 1076–1085.
- Wallis S, Lloyd S, Wise I, Ireland G, Fleming TP, Garrod D (2000). The alpha isoform of protein kinase C is involved in signaling the response of desmosomes to wounding in cultured epithelial cells. Mol Biol Cell. 11: 1077–1092.
- Wan H, South AP, Hart IR (2007). Increased keratinocyte proliferation initiated through downregulation of desmoplakin by RNA interference. Exp Cell Res. 313: 2336–2344.
- Wang H, Li Z, Yumul R, Lara S, Hemminki A, Fender P, Lieber A (2011a). Multimerization of adenovirus serotype 3 fiber knob domains is required for efficient binding of virus to desmoglein 2 and subsequent opening of epithelial junctions. J Virol. 85: 6390–6402.
- Wang H, Li ZY, Liu Y, Persson J, Beyer I, Moller T, Koyuncu D, Drescher MR, Strauss R, Zhang XB, Wahl JK, 3rd, Urban N, Drescher C, Hemminki A, Fender P, Lieber A (2011b). Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat Med. 17: 96–104.
- Wang H, Yumul R, Cao H, Ran L, Fan X, Richter M, Epstein F, Gralow J, Zubieta C, Fender P, Lieber A (2013). Structural and functional studies on the interaction of adenovirus fiber knobs and desmoglein 2. J Virol. 87: 11346–11362.
- Waschke J (2008). The desmosome and pemphigus. Histochem Cell Biol. 130: 21–54.
- Whittock NV, Ashton GH, Dopping-Hepenstal PJ, Gratian MJ, Keane FM, Eady RA, McGrath JA (1999). Striate palmoplantar keratoderma resulting from desmoplakin haploinsufficiency. J Invest Dermatol. 113: 940–946.
- Whittock NV, Wan H, Morley SM, Garzon MC, Kristal L, Hyde P, Mclean WH, Pulkkinen L, Uitto J, Christiano AM, Eady RA, McGrath JA (2002). Compound heterozygosity for non-sense and mis-sense mutations in desmoplakin underlies skin fragility/woolly hair syndrome. J Invest Dermatol. 118: 232–238.
- Williamson L, Raess NA, Caldelari R, Zakher A, de Bruin A, Posthaus H, Bolli R, Hunziker T, Suter MM, Muller EJ (2006). Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. EMBO J. 25: 3298–3309.
- Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, Pilichou K, Scherer SE, Saffitz J, Kravitz J, Zareba W, Danieli GA, Lorenzon A, Nava A, Bauce B, Thiene G, Basso C, Calkins H, Gear K, Marcus F, Towbin JA, Multidisciplinary Study of Right Ventricular Dysplasia Investigators (2010). Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 55: 587–597.
- Yamaji K, Fujimoto S, Ikeda Y, Masuda K, Nakamura S, Saito Y, Yutani C (2005). Apoptotic myocardial cell death in the setting of arrhythmogenic right ventricular cardiomyopathy. Acta Cardiol. 60: 465–470.
- Yang L, Chen Y, Cui T, Knosel T, Zhang Q, Albring KF, Huber O, Petersen I (20120.Desmoplakin acts as a tumor suppressor by inhibition of the Wnt/beta-catenin signaling pathway in human lung cancer.Carcinogenesis. 33: 1863–1870.
- Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, Ge S, Nadvoretskiy VV, Defreitas G, Carabello B, Brandon LI, Godsel LM, Green KJ, Saffitz JE, Li H, Danieli GA, Calkins H, Marcus F, Towbin JA (2006). Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res. 99: 646–655.
- Yin T, Getsios S, Caldelari R, Kowalczyk AP, Muller EJ, Jones JC, Green KJ (2005). Plakoglobin suppresses keratinocyte motility through both cell–cell adhesion-dependent and -independent mechanisms. Proc Natl Acad Sci U S A. 102: 5420–5425.
- Yoshida M, Romberger DJ, Illig MG, Takizawa H, Sacco O, Spurzem JR, Sisson JH, Rennard SI, Beckmann JD (1992). Transforming growth factor-beta stimulates the expression of desmosomal proteins in bronchial epithelial cells. Am J Respir Cell Mol Biol. 6: 439–445.
- Zhang M, Tavora F, Oliveira JB, Li L, Franco M, Fowler D, Zhao Z, Burke A (2012). PKP2 mutations in sudden death from arrhythmogenic right ventricular cardiomyopathy (ARVC) and sudden unexpected death with negative autopsy (SUDNA). Circ J. 76: 189–194.
- Zhang S, Chen D, Huang C, Bao J, Wang Z (2013). Expression of HGF, MMP-9 and TGF-beta1 in the CSF and cerebral tissue of adult rats with hydrocephalus. Int J Neurosci. 123: 392–399.
- Zhurinsky J, Shtutman M, Ben-Ze'ev A (2000a). Differential mechanisms of LEF/TCF family-dependent transcriptional activation by beta-catenin and plakoglobin. Mol Cell Biol. 20: 4238–4252.
- Zhurinsky J, Shtutman M, Ben-Ze'ev A (2000b). Plakoglobin and beta-catenin: protein interactions, regulation and biological roles. J Cell Sci. 113 (Pt 18): 3127–3139.