Abstract
Autism is dramatically increasing in incidence and is now considered an epidemic. There are no objective means to diagnose the disorder. Diagnosis is made subjectively, based on the perceived behavior of the subject. This review presents an approach toward development of an objective measure of autism. Covering the literature from 1943 to the present in the PubMed and Ovid Medline databases, this review summarizes evidence of hormones, metabolites, amino acids, and other biomarkers present in significantly different quantities in autistic subjects compared to age- and sex-matched controls. These differences can be measured in the gastrointestinal, immunologic, neurologic, and toxicologic systems of the body, with some biomarkers showing ubiquitous application. In addition, there are unifying concepts, i.e., increased vulnerability to oxidative stress, immune glutamatergic dysfunction, and pineal gland malfunction. The variances of the biomarkers from the norm present the opportunity to create biomarker arrays that when properly developed and analyzed could result in an objective diagnosis with a ranking of the severity of autism for each subject. The contribution of each biomarker to the overall diagnosis could be calculated, thus providing a profile pattern unique to the individual. This profile could consequently provide information for therapeutic interventions on an individual basis.
Introduction
Autism begins in infancy or, at the latest, in the first 3 years of life (Kolvin, Citation1971). Autism is a heterogeneous condition, but difficulties fall into areas that are reliably measured and usually consistent across time, even though specific behaviors may change with development. Children with autism are unable to interpret the emotional states of others, failing to recognize anger, sorrow or manipulative intent. Their language skills are often limited, and they find it difficult to initiate or sustain conversations. They frequently exhibit an intense preoccupation with a single subject, activity or gesture. Autism has many suspected causes; these are described in a companion manuscript submitted to Journal of Immunotoxicology (Ratajczak, Citation2010). There remains no objective means to diagnose this disorder even though autism was once stated to be the complex psychiatric or developmental disorder with the best empirically based, cross-national diagnostic criteria (Volkmar and Klin, Citation2005). The diagnosis of autism is done subjectively, and is thus dependent on the expertise of those administering the tests. Using subjective tests is an oversimplification that does not account for the complex interactions and manifestations of hormones, metabolites, and other biomarkers present in differing amounts in autistic patients. “The problem besetting all attempts to produce reliable and valid diagnostic categories for autism…is the continuing lack of any independent biological or psychological markers” (Wing, Citation2005). No biological markers have been found to reliably diagnose autism in an individual patient (Posey et al., Citation2008; Anderson, Citation2010).
With a wide variety of theoretical causes and known comorbidities, autism is very complex. It is imperative to identify biomarkers that are expressed differently in autistic patients. Because many autistic individuals have comorbidities, such as epilepsy or mental retardation, many biomarkers will overlap autism and the other disorders. However, the quantitation of the biomarker may be different in the two. Measurement of the biomarkers will provide a means of an objective diagnosis of autism and perhaps aid in definition of the comorbidities as well.
In addition to the subjective behavior measure, autistic subjects have been documented to have aberrances in hormones, peptides, metabolites, etc. from the neurologic, gastrointestinal (GI), immunologic, and toxicologic systems (Jepson, Citation2007a). Although, to date, no individual biomarkers can reliably be used to diagnose autism, it is possible these variances from the norm can provide biomarker arrays that, when properly developed and analyzed, might be diagnostic. Additionally, biomarker profile patterns could provide information for therapeutic interventions.
The biomarkers cited in the following sections have all been reported in peer-reviewed literature to be statistically significant compared to neurotypical age- and sex-matched controls. In some cases, other controls with disabilities such as mental retardation or epilepsy were also used.
Neuropathologies in autism
Biochemical and pathologic signs of autism present very early in life: Newborns with increased neurotransmitters have been shown to develop autism at a later age (Nelson et al., Citation2001). The neuropathology of infantile autism has its origins in the prenatal development of the brain, with an ongoing pathological process that continues into adult life (Kemper and Bauman, Citation1998). Consistent abnormalities in the limbic forebrain have given the strongest correlation with the clinical features of the disorder. Consistent findings have been seen in the limbic system, cerebellum, and the closely-associated inferior olive (located on anterior surface of the medulla), which are involved in control and coordination of movements, sensory processing and cognitive tasks (Wikipedia, Citation2010). In the limbic system, the hippocampus, amygdala, and entorhinal cortex have shown small cell size and increased cell-packing density in autistic patients of all ages, suggesting development curtailment. In the cerebellum there are significantly reduced numbers of Purkinje cells, primarily in the posterior inferior regions of the hemispheres. A different pattern of change was noted in the vertical limb of the diagonal band of broca, cerebellar nuclei and inferior olive with plentiful and abnormally enlarged neurons in the brains of young autistic subjects, and in adult autistic patients’ brains, with small, pale neurons that are reduced in number.
Together with age-related changes in brain weight and volume, it is possible that the neuropathology of autism represents an ongoing process (Bauman and Kemper, Citation2005). Consistent with this theory, the quantity of some biomarkers changes as the individuals increase in age (Chugani et al., Citation1999). Many markers are primarily found in the body’s four basic systems (GI, immunologic, neurologic, and Toxicologic) described by Jepson (Citation2007a).
Candidates for biomarkers of autism
GI
It has been reported that autistic children frequently have GI difficulties, including diarrhea, constipation, gaseousness, bloating, abdominal pain, stool impaction, reflux, and belching (Horvath et al., Citation1999). Several years later, reflux esophagitis was the most frequent finding of children with autism who underwent upper GI endoscopy. The clinical symptoms correlated with the histology. Of those with esophagitis due to reflux, 93% had at least one of the symptoms of abdominal pain, nighttime waking, and sudden daytime irritability. There are also functional GI abnormalities in autistic patients, particularly low activities of disaccharidase enzymes, e.g., lactase, maltase, sucrase, palatinase, and glucoamylase (Horvath et al., Citation1999; Horvath and Perman, Citation2002).
The clinical symptoms of GI involvement in autistic subjects are correlated with histology. Chronic gastritis was common with increased numbers of lymphoid aggregates and lymphocytic infiltrates in the mucosa. Histology revealed increased staining at the base of crypts where Paneth cells are localized. Autistic patients had elevated numbers of Paneth cells per crypt compared with controls. In addition, immunohistochemical studies showed a much higher lysozyme content in the Paneth cells of autistic subjects (Horvath and Perman, Citation2002). Previously, other investigators demonstrated a pattern of colitis and ileal-lymphoid-nodular hyperplasia in children with autism (Wakefield et al., Citation1998).
Porphyrins, derivatives of the heme synthesis pathway and measures of xenobiotic exposures, have been documented to be found at increased levels in the urine of autistic patients (Geier and Geier, Citation2006a; Nataf et al., Citation2006). In addition, a calcitonin gene-related peptide was reported to be increased in the blood of autistic subjects (Nelson et al., Citation2001). Another study of urine from autistic patients documented increased levels of a number of peptides, e.g., Deltophin II, Dermorphin, and Desmorphin (opioid-like peptides), a morphine modulating peptide, and Novel Autism Peptides I and III (Shanahan et al., Citation2000). In contrast, dipeptidyl peptidase IV (which is CD26 on T-lymphocytes), the only known enzyme to break down casomorphine, was very low or absent in the urine of autistic individuals, but normal in neurotypical controls. The absence of dipeptidyl peptidase IV could explain the opioid-like casomorphine found to be elevated in urine from autistic patients (Reichelt and Liu, Citation1997). Another opioid-like peptide, sauvagine, was also increased in the urine of autistic subjects (Mehl-Madrona, Citation2000). It is hypothesized that these opioids inhibit the sensation of pain in autistic subjects (Reichelt and Knivsberg, Citation2003).
The main idea of the opioid-excess theory suggests that excessive levels of incompletely metabolized peptides from foods that contain proteins, gluten and casein, pass through the intestinal and blood-brain barriers into the brain, where they directly regulate transmission in all the main neurotransmission systems or form ligands for peptidase enzymes that would normally hydrolyze naturally-occurring opioid peptides (Shattock and Whiteley, Citation2002). Panksepp (Citation1979) first proposed the opioid-excess theory of autism, reporting similarities between autism symptoms and effects of morphine, e.g., reduced social contact, insistence on sameness, decreased pain sensitivity, and delay in developmental milestones. According to the opioid hypothesis for autism, hyperfunction of the endogenous opioid system could explain most of the symptoms associated with autistic disorder (Gillberg, Citation1995). Increased amounts of the endogenous opioid β-endorphin are also found in autistic patients (Cazzullo et al., Citation1999), along with other endogenous peptides (measured in neonatal blood spots) such as vasoactive intestinal peptide, calcitonin gene-related peptide, brain-derived neurotrophic factor (BDNF), and neurotrophin 4/5 (Nelson et al., Citation2001).
In contrast, more recent studies have not found evidence of opioid peptides in the urine from autistic children or young adults (Hunter et al., Citation2003; Cass et al., Citation2008). It is possible that the cause of the opioids found in the older studies was no longer present in the individuals studied by Hunter et al. and Cass et al. The conflicting results strongly indicate the need for more research to understand how the endogenous opioid system interacts with neurotransmitters with respect to autism.
A compilation of some of the theoretical biomarkers of autism that pertain to the GI system and that were described in this section are provided in .
Table 1. Theoretical biomarkers of autism: gastrointestinal system.
Immunologic
Autoimmune responses are increased in autism. Levels of antibodies directed against autologous cerebellar peptides, dipeptidyl peptidase IV, and/or gliadin were increased in the serum of autistic subjects (Vojdani et al., Citation2004a, Citation2004b). Serum or plasma levels of other autoantibodies including those against glial filament and neurofilament proteins (Singh et al., Citation1997), and myelin basic protein were also increased (Singh et al., Citation1993). Connolly et al. (Citation2006) documented increased autoantibody levels against BDNF, brain endothelial cells, and myelin basic protein in the sera of autistic children.
Levels of immunoglobulin A have also been documented to be low (Warren et al., Citation1997), but those of cytokines interleukin (IL)-1β and IL-6 were elevated (Jyonouchi et al., Citation2001) in the mononuclear blood cell cultures of autistic patients. Other studies have also reported elevated levels of tumor necrosis factor (TNF)-α (Jyonouchi et al., Citation2001), soluble IL-2, and soluble CD8 (Singh et al., Citation1991), as well as IL-12 and interferon (IFN)-γ (Singh, Citation1996).
Vargas et al. (Citation2005) studied the brain tissues from deceased patients with autism for concentrations of cytokines. Eighteen cytokines were quantitated in the middle frontal gyrus, anterior cingulated gyrus, and/or cerebellum, with significant increases compared to controls. IL-6 had the greatest increase, at 31.4-fold in the anterior cingulated gyrus. Cytokines were also quantitated in the cerebrospinal fluid (CSF) of autistic individuals, with the peak increase of 232.5-fold for IFNγ.
It should be noted that in the study of Vargas et al. (Citation2005) there was a lack of adaptive immune reactions in the brain of patients with autism. A few isolated perivascular CD3+ and CD20+ cells in both autistic and control brains were identified, but there was no evidence of leptomeningeal, parenchymal, or perivascular inflammatory infiltration in autistic subjects’ brains. Immunostaining with antibodies specific for immunoglobulin G (IgG), IgA, or IgM showed no deposition of these immunoglobulins in neuronal or neuroglial cell populations. There was deposition of complement membrane attack complexes in the cerebella of autistic brains. The absence of adaptive immune reactions in these brains demonstrate that the autoimmune antibodies documented above (Singh et al., Citation1993, Citation1997; Vojdani et al, Citation2004a,Citationb), if present in the subjects studied, did not cross the blood-brain barrier or that lack of a complement component prevented the normal sequela of the interaction of antibodies with their specific antigens. The data suggest that the adaptive immune system does not play a significant pathogenic role, with the main immune mechanism being innate reactions.
The cerebellum has been a main focus of neuroinflammation in autism (Vargas et al., Citation2005). The most prominent cytokines in the brain of autistic patients were macrophage chemoattractant protein (MCP)-1 and tumor growth factor-β1. There was a marked expression of proinflammatory cytokines in the CSF of autistic patients. MCP-1, considered one of the most relevant proteins because it was significantly elevated in both brain tissues and the CSF, facilitates the infiltration and accumulation of monocytes and macrophages in inflammatory central nervous system disease.
C4B complement protein was found to be deficient in the blood of autistic patients (Warren et al., Citation1994, Citation1995). This finding correlates with studies that indicate the complement C4B gene null allele (i.e., the missing or nonfunctional C4B gene) is more frequent in individuals with autism (Yonk et al., Citation1990; Warren et al., Citation1991). The C4B protein is essential in the activation of the classical complement pathway that results in lysis (Kunkel et al., Citation1985). C4B is considered a regulatory protein, and covalently binds to the surface of target cells. In the absence of C4B, the immune system is partially compromised.
Confirming the association of the C4B null allele with autism is the fact that there is an increased incidence of the C4B null allele in attention-deficit hyperactivity disorder and reading disability (Warren et al., Citation1996). In addition, the DR molecules of the major histocompatability complex (MHC) are composed of chains with polymorphic amino acid substitutions clustered in three hypervariable regions (HVRs), necessary for the development of specific cell-mediated and humoral immune responses to pathogens and other foreign antigens. The HVR-3 sequence has significantly elevated frequency in subjects with autism, and is also associated with rheumatoid arthritis, an autoimmune disease (Warren et al., Citation1996). There is an elevated incidence of immune and autoimmune disorders in autistic patients and in their first-degree relatives (Megson, Citation2000).
Other abnormal immune parameters in autism reported were lymphocyte subsets, with helper (CD4+) T-lymphocytes documented as deficient (Warren et al., Citation1990; Denney et al., Citation1996). Natural killer cell function was also decreased (Warren et al., Citation1987). Inversely correlated with symptoms of autism were the helper:suppressor (CD4:CD8) ratio. Autistic individuals had fewer lymphocytes with bound IL-2 on their surface following mitogenic stimulation. Alternately, DR+ activated T-lymphocytes were increased in blood of autistic patients in contrast to normal IL-2 receptors on lymphocytes, suggesting incomplete activation of the cells, often seen in autoimmune disease (Plioplys et al., Citation1994). DR+ T-lymphocytes without the IL-2 receptor were also detected in autistic individuals, and the DR+ T-lymphocytes were inversely correlated with a decreased plasma level of the C4B protein (Warren et al., Citation1994, Citation1995). The B-lymphocyte antigen D8/17, with expanded expression in rheumatic fever, Sydenham’s chorea, and subgroups of obsessive-compulsive disorder and Tourette’s syndrome with repetitive behaviors, was reported to be expressed to a greater degree in children with autism, and was correlated with the severity of repetitive behaviors (Hollander et al., Citation1999). Eosinophils were elevated in the blood of autistic subjects (Renzoni et al., Citation1995) and the monocyte count was elevated (Denney et al., Citation1996; Sweeten et al., Citation2003).
The immune function of cells in autistic patients was also reported to be deficient. Lymphocyte proliferation to mitogens was decreased in autistic individuals compared to that by cells from neurotypical controls (Stubbs and Crawford, Citation1977; Warren et al., Citation1986). Historically, there is confirmation of an alteration in migration inhibition factor (MIF) data in autism. In 1982, production of MIF by mononuclear white blood cells in response to myelin basic protein was increased in autistic subjects (Weizman et al., Citation1982). Over 20 years later, autistic patients had increased amounts of circulating MIF in their blood (Grigorenko et al., Citation2008). Moreover, the level of circulating MIF was correlated with behavioral indicators measured by Autism Diagnostic Observational Schedule scores on social impairment, imaginative skills, and total score.
Consistent with immune panels of many autistic children revealing signs of atypical infections and shifted cell counts, it is documented that subgroups of autism display intra-monocyte pathogens, such as measles virus, cytomegalovirus-virus, human herpes virus 6 and Yesinia enterocolitica, with manifestations of lowered hematopoiesis, impaired peripheral immunity, and altered blood-brain barrier functions often accompanied by demyelination (Binstock, Citation2001).
These changes in immune function may be caused by serotonin, which has been shown to influence immune parameters such as T-lymphocyte subpopulations, lymphocyte transformation, and macrophage accessory function (Slauson et al., Citation1984; Young and Matthews, Citation1995; Mössner and Lesch, Citation1998; Stefulj et al., Citation2001; Burgess et al., Citation2006).
Immunologic changes in autism include production of autoantibodies to proteins in the brain, low levels of IgA, and increased inflammatory cytokines, not only in the blood, but in the brain and CSF as well. In addition, some autistic individuals have lack of the C4B complement protein, increased HVRs in the MHC, deficient helper T-lymphocytes, abnormal helper:suppressor lymphocyte ratios, incomplete activation of T-lymphocytes, increased expression of the B-lymphocyte antigen D8/17, increased numbers of eosinophils and monocytes, altered immune function (decreased proliferation of lymphoyctes in response to mitogens), and intra-monocyte pathogens. The data support the role of autoimmunity in autism, with innate immune responses being the effectors.
A compilation of some of the theoretical biomarkers of autism that pertain to the immune system and that were described in this section are provided in .
Table 2. Theoretical biomarkers of autism: immunologic system.
Neurologic
To date, the strongest evidence implicates systems involved with glutamate, gamma-amino butyric acid (GABA) and serotonin systems (with melatonin as a major effector) in autism, with weaker evidence for changes in catecholaminergic, peptidergic, and cholinergic systems (Polleux and Lauder, Citation2004). There are contrasting hypotheses about autism and glutamate, with some investigators saying glutamate is decreased (Carlsson, Citation1998) and others saying it is increased, possibly due to a decreased level of inhibition (Rubenstein and Merzenich, Citation2003). Moreno et al. (Citation1992) documented that glutamate was increased in the plasma of autistic individuals. These data support the hypothesis that glutamate is increased in autism.
An abnormality in the GABA system in autism was first documented by Blatt et al. (Citation2001), who used autoradiography to prove that the density and distribution of hippocampal neurotransmitter receptors in the GABA receptor system is significantly reduced in high-binding regions. The suppressed GABA inhibition may contribute to the pathophysiology of autism (Belmonte et al., Citation2004). GABA has several subunits of its receptor. Studies in rats show GABA to be highly expressed during brain development (Laurie et al., Citation1992). Other research showed that knockout mice lacking the GABRB3 gene display seizures, hypersensitive behavior, learning and memory deficits, poor motor skills on a repetitive task, hyperactivity, and a disturbed rest-activity cycle, features common to the Angelman syndrome in humans (DeLorey et al., Citation1998). The Angelman syndrome has some similarities to autism (Belmonte et al., Citation2004).
The expression of the α4β2 nicotinic acetylcholine receptor was decreased in autopsied brains of adults with autism (Perry et al., Citation2001; Lee et al., Citation2002). In contrast, there were normal levels of choline acetyltransferase, suggesting that the cholinergic input to the cerebral cortex and cerebellum was intact. However, the level of BDNF in the basal forebrain was three times normal (Lee et al., Citation2002). It should be noted that BDNF measured in blood spots from neonates (Nelson et al., Citation2001) and serum from children (Connolly et al., Citation2006) was increased in autistic individuals. In contrast, Hashimoto et al. (Citation2006) found BDNF to be decreased in serum of autistic adults. BDNF is involved in enhancing cholinergic transmission and in promoting the survival of developing cholinergic neurons of the basal forebrain (Ward and Hagg, Citation2000). Thus, pharmacologic therapies targeting the cholinergic system may be of value. A study supporting this is the successful use of the cholinesterase inhibitor Donezipil to treat autistic patients’ symptoms of irritability and hyperactivity (Hardan and Handen, Citation2002).
Platelet hyperserotonemia is generally considered the most robust and well-replicated biological finding in autism (Belmonte et al., Citation2004) and the increase in this neurotransmitter has been documented by many studies, including Martineau et al. (Citation1992), Rolf et al. (Citation1993), Chugani et al. (Citation1999), and Leboyer et al. (Citation1999). Serotonin plays a key role in behaviors and processes, including sleep, mood, arousal, aggression, impulsivity, and affiliation (Lucki, Citation1998). In the embryo, serotonin acts as a growth factor and regulator of neuronal development (Whitaker-Azmitia, Citation2001). The serotonergic system is intimately interconnected with GABAergic and glutamatergic neurons throughout the brain. Serotonin is a precursor of melatonin, converted to N-acetylserotonin in the pineal gland by the rate-limiting enzyme arylalkylamine N-acetyltransferase, and followed by the conversion of N-acetylserotonin to melatonin by acetylserotonin methyltransferase (ASMT). There is also a significant negative correlation between testosterone and whole blood serotonin in autistic patients (Tordjman et al., Citation1995) (see below).
The pineal gland, located in the exact center of the brain, is considered to be a functioning neuroendocrine transducer, an organ that converts a neural signal conveying environmental information into an endocrine message (Axelrod, Citation1974; Brainard, Citation1978). Thus, the pineal gland plays a key role in the development of the brain and the hormonal system. The pineal gland functions during the dark, and is inhibited in the light. Since 1920, pineal gland extracts have been used in the treatment of mental illnesses (Kitay and Altschule, Citation1954; Brainard, Citation1978). The primary hormonal agent of the pineal gland is N-acetyl-5-methoxy-tryptamine (melatonin), which was only discovered after 40 years of investigation (Lerner, et al., Citation1958, Citation1960).
Melatonin is decreased in the urine, saliva, and blood of autistic patients (Nir et al., Citation1995; Axt, Citation1996; Kulman et al., Citation2000; Melke et al., Citation2008). The most notable feature of melatonin is that its production normally follows a circadian rhythm, with peak production at night (Reiter and Robinson, Citation1995a). In autism, the quantity of melatonin is below normal, and a circadian rhythm in its production is not displayed (Kulman et al., Citation2000; Melke et al., Citation2008). It is activation of receptors in the pineal gland that begins the enzymatic process that leads to synthesis of melatonin, the main pineal secretion. Melatonin is synthesized from dietary l-tryptophan (Axt, Citation1996), with the enzymes involved in the synthesis activated and depressed by darkness and light, respectively ().
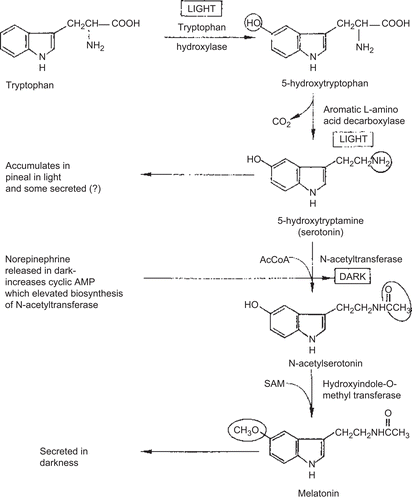
Testosterone is increased in serum of autistic individuals (Geier and Geier, Citation2006b), and there is an effect of testosterone on melatonin secretion. Decreased nocturnal secretion of melatonin has been documented in patients with Klinefelter’s syndrome (Luboshitzky et al., Citation1996), the most common cause of primary hypogonadism in men, resulting in impairment of both sperm and androgen production. Male patients with low testosterone hypergonadotrophic hypogonadism had decreased melatonin levels, in contrast to Klinefelter’s patients with normal testosterone in which melatonin levels were normal. Male patients with low levels of gonadotropin-releasing hormone (GnRH) have increased nocturnal melatonin secretion; this is in contrast to hypergonadotrophic hypogonadal males who display decreased melatonin secretion (Luboshitzky et al., Citation1997). Testosterone treatment normalized melatonin concentrations in both groups of patients. The results suggest that GnRH, gonadotrophins, and gonadal steroids modulate pineal melatonin in humans. Fetal testosterone has been linked with autism (Knickmeyer et al., Citation2005; Auyeung et al., Citation2009). It is possible this early influence of testosterone on the developing fetus is responsible for the lower level of melatonin in these patients. To date, no definitive studies have addressed testosterone-lowering medication for the treatment of autism.
Other hormones are also implicated in the etiology of autism. Norepinephrine and epinephrine are decreased in the urine of autistic individuals (Martineau et al., Citation1992). Blood levels of adrenocorticotropin hormone (ACTH) were increased in autistic subjects (Curin et al., Citation2003). In contrast, cortisol was decreased. However, cortisol was increased in autistic children in anticipation of reexposure to a perceived stressor (Corbett et al., Citation2006, Citation2008). A slower elevation of cortisol after ACTH stimulation was documented for autistic individuals compared to healthy controls. These autistic individuals did not display differences in salivary cortisol circadian rhythm or daily cortisol excretion (Marinovic-Curin et al., Citation2008).
Neuropathological abnormalities have been reported in cholinergic nuclei in the basal forebrain of autistic individuals (Bauman and Kemper, Citation1994, Citation2005). Because cholinergic afferents innervate the cerebral cortex during the most dynamic periods of neuronal differentiation and synapse formation, suggesting they play a regulatory role in these events (Hohmann and Berger-Sweeney, Citation1998), measures of cholinergic transmitter activity were investigated (Perry et al., Citation2001). Cholinergic biomarkers were measured in the basal forebrain and cerebral cortex in autopsy tissue from autistic adults. The level of BDNF was three times as high in the autistic subjects’ brains compared to the normal controls. Although choline acetyltransferase levels were found to be normal, many nicotinic (high-affinity) and moderate muscarinic M1 receptor measures in the cerebral cortex were lower than normal. The muscarinic receptor-binding abnormality reflected a low number of receptors. This could be related to epilepsy, since a low number has been reported in hippocampal sclerosis associated with temporal lobe epilepsy (Pennell et al., Citation1999).
The lower degree of high-affinity nicotinic receptor binding in the cortex of the autistic subjects was extensive, and reflects low receptor numbers (Perry et al., Citation2001). There was lower epibatidine binding, consistent with the preliminary findings of lower immunoreactivity of the nicotinic α4 and β2 subunits. Low nicotinic receptor binding and synaptophysin have been identified in Alzheimer’s disease (Sabbagh et al., Citation1998), suggesting that the low receptor binding in autism may be associated with abnormal cortical neuronal morphology, possibly involving over-extensive synaptic pruning. Others confirmed that expression of the α4β2 nicotinic acetylcholine receptor is decreased in the autopsied brains of autistic adults (Lee et al., Citation2002). Nicotinic agents may be analgesic without the problems associated with opioids, such as causing a decrease in gastric motility (Bannon et al., Citation1998).
Because the α4 nicotinic subunit has been implicated in pain perception, low levels of this receptor subtype in autism may be associated with the low degree of pain reactivity that is present in the disorder (Tordjman et al., Citation1999). Levels of β-endorphin and ACTH are greater in autistic subjects than normal individuals (Tordjman et al., Citation1997). More recent work on pain perception in autism has demonstrated that in response to venipuncture, there is a greater heart rate response and elevated plasma β-endorphin levels in autism subjects compared to age-, sex-, and pubertal stage-matched controls (Tordjman et al., Citation2009). The results strongly suggest that prior reports of reduced pain sensitivity in autism are related to a different mode of pain expression rather than to an insensitivity or endogenous analgesia, and do not support opioid theories of autism.
In a study of molecular analysis of nicotinic receptor expression, α4 mRNA levels, α4 and β2 protein expression and receptor-binding density were lower in the parietal cortex in the brains of adults with autism (Martin-Ruiz et al., Citation2004). In the cerebellum of these brains, α4 mRNA expression was increased, but subunit protein and receptor levels were decreased. In contrast, α7 receptor binding in the cerebellum was increased. These data confirm those of Perry et al. (Citation2001). Retinoid receptors in the brain are also involved in autism: there is a disconnect between G-alpha protein and retinoid receptors caused by the pertussis toxin found in the DPT vaccine in genetically at-risk children (Megson, Citation2000).
A defect of cholesterol biosynthesis was found to cause the Smith-Lemli-Opitz syndrome (SLOS) (Tint et al., Citation1994). SLOS manifests some anatomical abnormalities reminiscent of those often seen in autism: hypoplasia of the corpus callosum, the cerebellum, and particularly, the vermis (Kelley and Hennekam, Citation2000). In autism, the developing macrocephaly contrasts with the microcephaly in SLOS present from birth and persisting into later life. Clinical data show that SLOS is associated with autism (comorbidity) (Lord et al., Citation1994), and suggest that cholesterol supplementation ameliorates autistic subjects’ behavioral symptoms (Tierney et al., Citation2001).
Biopterin and neopterin were increased in the urine of 3–5-year-old autistic children (Messahel et al., Citation1998). Similarly, neopterin was increased in the plasma of autistic children (Sweeten et al., Citation2003). The pterins’ synthesis starts from GTP, metabolized to 7,8-dihydroneopterin triphosphate by GTP cyclohydrolase, which is further metabolized to 5,6,7,8-tetrahydrobiopterin (BH4) or neopterin. Neopterin is associated with activation of cell-mediated immunity, and is a marker of disease activity for autoimmunity. Neopterin accompanies the immune activation of macrophages; both IFNγ and TNFα cause increased amounts of neopterin to be produced. Neopterin levels are raised significantly during exacerbation of autoimmune diseases, and are lower during remission, thus acting as a marker of disease activity (Fuchs et al., Citation1993). Due to an increase in BH4 biosynthesis, levels of biopterin were also found to be increased in autistic patients (Messahel et al., Citation1998). IFNγ causes many types of cells to make BH4; any one of these cells could be the source of biopterin (Werner et al., Citation1989). BH4 acts as a cofactor for various reactions, including NO synthetase and the aromatic amino acid hydroxylases; it is also required for the biosynthesis of the neurotransmitters dopamine, adrenalin, and serotonin (Kaufman, Citation1986). Any disruption in BH4 metabolism can lead to neurological dysfunction.
The levels of several neuropeptides and neurotrophins were elevated in the neonatal (age 38.7 weeks) blood of children with autism, compared to neurotypical age-matched controls (age 38.4 weeks) and to children with cerebral palsy (age 38.4 weeks). These included vasointestinal peptide, neurotrophin 4/5, calcitonin gene-related peptide, and BDNF. However, the levels of these markers were not different from those of children with mental retardation without autism (Nelson et al., Citation2001). In contrast, concentrations of Substance P, pituitary adenylate cyclase-activating polypeptide, nerve growth factor, and neurotrophin 3 did not differ by diagnostic group. The substances investigated play major roles in the biology of brain development. The use of blood drawn in the newborn period had the advantage that sampling was relatively early in the sequence of pathogenic events and was not confounded by effects of treatment. An association study between BDNF gene polymorphisms and autism by three-dimensional gel-based microarray documented that a single nucleotide polymorphism of BDNF displayed a significant difference in the frequency of the C270T polymorphism and genotype, and there was a significant difference in global haplotype distribution between autistic subjects and matched controls (Cheng et al., Citation2009). The data suggest that BDNF has a possible role in the pathogenesis of autism.
Neurotrophins have multiple functions during peripheral and central nervous system development, such as regulating the growth, development, survival and repair of the nervous system (Kaplan and Miller, Citation2000). BDNF is a major neurotrophin, and plays multiple roles during neuronal differentiation, including neuronal survival, activity-dependent dendritic and axonal outgrowth/branching, synapse formation, and neuronal plasticity underlying learning and memory (Polleux and Lauder, Citation2004). A possible cause of the increased BDNF in autism is mutations in the MeCP2 gene, which encodes a protein that has been proposed to function as a global transcriptional repressor. Mutations in MeCP2 cause Rett syndrome, which is associated with autism. MeCP2 has been shown to bind selectively to a BDNF promoter and functions to repress expression of the BDNF gene (Chen et al., Citation2003). MeCP2 plays a key role in the control of neuronal activity-dependent gene regulation and deregulation of this process may underlie the pathology of Rett syndrome. There is overlap between Rett syndrome (which is found primarily in girls) and autism (primarily occurring in boys) (Muhle et al., Citation2004).
A compilation of some of the theoretical biomarkers of autism that pertain to the nervous system and that were described in this section are provided in .
Table 3. Theoretical biomarkers of autism: neurologic system.
Toxicologic
The liver is the main organ of detoxification, using different metabolic pathways to safely excrete harmful substances via the urine or the stool. In the liver, the main detoxification pathways are glucuronidation and transsulfuration. The brain is particularly sensitive to damage from toxins, and has an additional level of protection, the blood-brain barrier, which prevents many harmful substances from gaining access. However, the brain needs glucose and oxygen, and certain small toxic molecules are able to get into the brain. Some substances, such as free radicals, are metabolic by-products of the oxidation process. Reduction converts the free radicals back into a harmless form. Ultimately, an imbalance between oxidation and reduction (redox) creates oxidative stress, which can result in tissue injury.
Glutathione (GSH) is the most important antioxidant utilized for the detoxification and elimination of environmental toxins (Chauhan and Chauhan, Citation2006). The levels of reduced GSH were decreased in the plasma of autistic subjects (James et al., Citation2004; Geier and Geier, Citation2006b; Geier et al., Citation2009), in contrast to those of oxidized glutathione (GSSG) that were increased. GSH plays a major role in methylation, a process that is intimately involved in detoxification.
The methylation cycle of autistic individuals has been shown to be aberrant in each of its steps, with the expression of most key markers being lower than those found in control children (James et al., Citation2004). These include: methionine, S-adenosylhomocysteine (SAM), homocysteine, cystathionine, cysteine, and total GSH (tGSH). In contrast, greater values were found for S-adenosylhomocysteine (SAH), adenosine, and oxidized glutathione (GSSH). The ratios of SAM:SAH and tGSH:GSSG were almost 50% lower in autistic subjects than in controls. The data may be best explained by oxidative inactivation of methionine synthase, in combination with a decrease in SAH hydrolase activity secondary to the increase in adenosine. A combined enzyme deficit would also be consistent with the observed decrease in SAM and increase in SAH levels. The decrease in homocysteine concentrations would reflect an adenosine-mediated decrease in SAH hydrolysis and homocysteine synthesis. The increase in GSSG disulfide and decrease in tGSH:GSSG indicate chronic oxidative stress.
A striking return to normalcy was achieved with targeted nutritional intervention in those individuals described above. For example, supplementation with folinic acid and betaine caused the mean concentrations of methionine, SAM, SAH, adenosine, and homocysteine to become indistinguishable from normal (James et al., Citation2004). However, the intervention did not normalize tGSH or GSSG levels or tGSH:GSSG ratios. The addition of ingestible methylcobalamin to the folinic acid and betaine supplementation further decreased the concentrations of adenosine and GSSH and increased the concentrations of methionine, cysteine, and tGSH, as well as the ratios of SAM:SAH and tGSH:GSSG.
The work of James et al. (Citation2004) has great relevance because it demonstrates that a significant abnormality in a critical metabolic pathway in children with autism can be returned to near normal using nutritional supplementation. The attending physician noted subjective clinical improvements in both speech and cognition, but they were not measured in a quantifiable manner. Most of the results of James et al. were confirmed (Geier and Geier, Citation2006b; Geier et al., Citation2009). In addition to the James group’s findings, free sulfate was documented to be lower in autistic subjects than controls (Geier et al., Citation2009). Considering the results cited above, and the fact that every component of the methylation pathway is present at abnormal quantities in autistic subjects, the pathway presents a list of biomarkers for autism.
Sulfate in the gut is part of the body’s first line of defense against toxicants, neutralizing various substances before they can be absorbed (Jepson, Citation2007b). The metabolism of cysteine is the largest source of sulfate in the body (Jepson, Citation2007c). Children with autism have low plasma sulfate levels (Waring and Klovrza, Citation2000; Geier et al., Citation2009). Abnormal sulfation has the strong potential of causing or contributing to many abnormalities commonly seen in autism, including leaky gut syndrome, detoxification abnormalities, and neurotransmitter dysfunction (Jepson, Citation2007d). The abnormal sulfation could be caused by the decreased amount of phenylsulphotrans-ferase in autistic children, which decreases the metabolism of phenolic foods known to create abnormal behavior in autistic children (Waring et al., Citation1997).
A compilation of some of the theoretical biomarkers of autism that pertain to toxicologic systems and that were described in this section are provided in .
Table 4. Theoretical biomarkers of autism: toxicologic system.
Ubiquitous biomarkers
Amino acids
Several biomarkers apply to the entire body, since the metabolism involved is ubiquitous. Amino acids documented to be decreased in the blood of autistic subjects include aspartic acid, glutamine, glutamic acid, and GABA (Rolf et al., Citation1993). Evans et al. (Citation2008) report significantly lower urinary levels of essential amino acids for both untreated autistic children (aged 5–15 years) and those treated for digestive function and nutritional uptake compared to controls, with the most obvious difference in levels of tryptophan. This confirms the work of Arnold et al. (Citation2003) who reported that plasma from children (< 5 years old) with autism had more essential amino acid deficiencies (including the neurotransmitter precursors tyrosine and tryptophan) that were consistent with poor protein nutrition. In contrast, Aldred et al. (Citation2003) reported that 4–29-year-old autistic subjects had elevated plasma levels of glutamic acid, phenylalanine, asparagines, tyrosine, alanine, and lysine, concurrent with reduced levels of glutamine. The levels of all the other amino acids were within normal range. These findings in autistic subjects were similar to those of patients with Asperger’s syndrome and siblings and parents of both types of patients. The discrepancies between the data of Aldred et al. (Citation2003) and Rolf et al. (Citation1993) could be caused by differences in body fluids analyzed and differences in age of the subjects.
Autistic individuals also exhibited decreases in the plasma levels of phospholipid fatty acids (Vancassel et al., Citation2001), and pyruvate in the serum (Filipek et al., Citation2004). Carnitine has also been determined to be decreased in the serum of autistic patients (Filipek et al., Citation2004). Carnitine is a betaine, which is an oxidation product of choline and a transmethylating intermediate in metabolism isolated from muscle and liver. Carnitine is an essential cofactor involved in the utilization of fat during fasting and stress, and plays a key role in transport of long chain fatty acids into mitochondria where they undergo β-oxidation in energy production. In contrast, the levels of alanine (α-aminopropionic acid; one of the amino acids occurring widely in proteins), along with ammonia, are increased in the serum of autistic patients (Filipek et al., Citation2004). The decrease in the levels of carnitine, coupled with significant elevations in alanine and ammonia levels in autistic individuals, suggests mitochondrial dysfunction (Filipek et al., Citation2004).
cAMP
Cyclic adenosine 3′5′monophosphate (cAMP) is a second messenger involved in many processes including mnemonic processing and anxiety. Since a variety of tissues contribute to plasma cAMP levels, the cAMP is not specific to brain (Ahloulay et al., Citation1996). Memory deficits and anxiety are noted in the phenotype of fragile X (FX), the most common heritable cause of mental retardation and autism (Kelley et al., Citation2008). Signaling deficiencies of cAMP are central to FX pathophysiology (Miyashiro and Eberwine, Citation2004). In contrast, plasma levels of cAMP are elevated in medicated and unmedicated autistic children (those without FX) relative to levels in controls (Hoshino et al., Citation1980).
A positive correlation was found between the plasma cAMP level and hyperkinesis and serum serotonin levels in autistic patients (Hoshino et al., Citation1980). Also, the production of cAMP by adenylate cyclase is decreased by opioids; this effect (i.e., opioids causing reductions in the production of cAMP) is increased in autism (Panksepp, Citation1979; Kelley et al., Citation2008). Several studies document that naltrexone (an opioid antagonist that improves adenylate cyclase function) is beneficial to some aspects of autism behavior in some patients (Riddle et al., Citation1999; Elchaar et al., Citation2006), but not to all behaviors (Willemsen-Swinkels et al., Citation1995; Feldman et al., Citation1999).
The opiate theory of autism, mentioned above, proposes that autistic individuals have a hyperactive opiate system. Opioids are known to reduce the ability of adenylate cyclase to produce cAMP. cAMP may be a useful biomarker to differentiate between FX and autism (Kelley et al., Citation2008). In addition, the cAMP cascade may be a viable therapeutic target for both FX and autism.
A compilation of some of the theoretical biomarkers of autism that pertain to ubiquitous biomarkers and that were described in this section are provided in .
Table 5. Theoretical biomarkers of autism: ubiquitous.
Unifying concepts
Increased vulnerability to oxidative stress
The association of autism with genetic deficits in specific enzymes exhibited by the Rett syndrome, FX, phenylketonurea, adenylosuccinate lyase deficiency, dihydropyrimidine dehydrogenase deficiency, and 5′-nucleotidase hyperactivity suggests the possibility that the genetic component of primary autism could be expressed as a chronic metabolic imbalance that impairs normal neurodevelopment and immunologic function (James et al., Citation2004). Investigating this possibility, James et al. documented that, relative to age- and sex-matched controls, children with autism had significantly lower plasma concentrations of methionine, SAH, homocysteine, cystathionine, cysteine, and total GSH, in conjunction with significantly higher concentrations of SAH, adenosine, and GSSG. This profile demonstrates impaired capacity for methylation and increased oxidative stress. To confirm this association, a nutritional intervention trial was effective in bringing all the metabolites within the methionine cycle into normal ranges and significantly improved the metabolites in the transsulfuration pathway. The components of the methionine cycle could serve as metabolic biomarkers for autism.
Immune glutamatergic dysfunction
Most heterogeneous symptoms of autism have a common set of events closely connected with dysregulation of glutamatergic neurotransmission in the brain with enhancement of excitatory receptor function by proinflammatory immune cytokines as the underlying mechanism (Blaylock and Strunecka, Citation2009). Environmental and dietary excitotoxins, mercury, fluoride, and aluminum, can exacerbate the pathological and clinical problems by worsening excitotoxicity and by microglial priming. In addition, each has effects on cell signaling that can affect neurodevelopment and neuronal function. This dysregulation might be caused by linkage and association of the glutamate receptor 6 (GluR6) gene with autism (Jamain et al., Citation2002). In addition, there is partial duplication of the metabotropic glutamate receptor 8 (GRM8) gene with a possible association with autism (Serajee et al., Citation2003). Glutamate is the principal excitatory neurotransmitter in the brain, acting at more than half of its synapses, and is directly involved in cognitive functions such as memory and learning (Jamain et al., Citation2002; Blaylock and Strunecka, Citation2009). Other genetic evidence, a high incidence of mutation of the GRM8 gene controlling the metabotropic GluR8 receptor subunit, which negatively modulates glutamate neurotransmission, was found in families having autistic children (Serajee et al., Citation2003).
A number of factors can trigger both the inflammatory cascade and the excitotoxic cascade, including aluminum and mercury (adjuvant and preservative, respectively), both found in vaccines. The activation of brain microglia can occur by way of macrophage/lymphocyte interactions and by IL-1β (a cytokine) infiltration into the brain, demonstrated by the effect of systemic infection on cognitive decline in Alzheimer’s disease (Holmes et al., Citation2003). The amygdala is regarded as a critical limbic site for integration and processing of autonomic, endocrine, and behavior-related information (Davis et al., Citation1994; Buller and Day, Citation2002). Considering the available information, a unifying concept for autism would be to measure glutamate and immune cytokines to characterize autism.
Pineal gland malfunction
The pineal gland is the first gland in the human body to be formed, clearly distinguishable 3-weeks after conception (Reiter and Robinson, Citation1995b). Melatonin has multiple life-giving roles in the body, as it: boosts the immune system, is the most potent antioxidant, protects against environmental hazards, helps maintain a healthy heart, helps to prevent cancer, and extends the lifespan (Reiter and Robinson, Citation1995c). Melatonin is also used therapeutically to treat sleep disturbances, endocrine and immune dysfunction, abnormal brain function, seasonal affective disorder, and abnormalities in the central nervous, GI, and renal systems (Malhotra et al., Citation2004). Melatonin is ubiquitously distributed and uses a variety of mechanisms to modulate the physiology and molecular biology of cells. By virtue of its ablity to detoxify free radicals and related oxygen derivatives, melatonin influences the molecular physiology of cells via receptor-independent means (Reiter et al., Citation2010). Melatonin has little or no toxicity, and is inexpensive and readily available. As mentioned above, the quantity of melatonin in autistic subjects is decreased compared to neurotypical age- and sex-matched controls. In addition, the melatonin in autistic individuals does not have a characteristic circadian rhythm.
Humans, like all other organisms that inhabit this earth, have a rhythmic order underlying life (Koukkari and Sothern, Citation2006). In fact, the absence of a rhythm, such as the beating of the heart or cessation of brain waves, defines death. Circadian rhythms are those that pulse at approximately each 24 h, or the period of a day. Two hormones displaying marker rhythms (those most prominent and consistent) of the human body are cortisol and melatonin, with peak production in the morning and night, respectively (Selmaoui and Touitou, Citation2003). When one hormone is out of synchronization, all the other hormones are affected, with a “feed sidewards” manifestation (Sanchez de la Pena, Citation1993). Cortisol (Curin et al., Citation2003) and melatonin (Nir et al., Citation1995; Tordjman et al., Citation2005; Hare et al., Citation2006; Melke et al., Citation2008) are both found to be deficient in autistic subjects, and to display circadian rhythms with dampened amplitudes (Nir et al., Citation1995; Hare et al., Citation2006). These deficiencies can have a severe effect on the body and its functions. For example, abnormal melatonin concentrations can have a dramatic effect on human behavior, as shown in patients with Smith–Magenis syndrome, who have an inverted melatonin circadian rhythm and display autistic-like behaviors (Melke et al., Citation2008).
A low melatonin level, shown to be caused by a primary deficit in ASMT, the enzyme which converts N-acetylserotonin to melatonin), has been documented as a risk factor for autism, and supports ASMT as a susceptibility gene for autism. Melatonin plays a crucial role in human cognition and behavior (Melke et al., Citation2008). In addition, an abnormal melatonin rhythm correlated with sleep studies in children with autism has been documented over the past four decades (Ornitz et al., Citation1965; Ornitz, Citation1973; DeMyer et al., Citation1981; Hoshino et al., Citation1982; Volkmar et al., Citation2004; Malow et al., Citation2006; Glickman, Citation2010). Melatonin has been used successfully in treatment of chronic sleep disorders in individuals with autism (Giannotti et al., Citation2006; Galli-Carminati et al., Citation2009; Wirojanan et al., Citation2009).
Summary and conclusion
The modifications described in this review of many theoretical biomarkers of autism have been summarized in . Biomarkers are listed in their primary system of the body (GI, immunologic, neurologic, and toxicologic, with some ubiquitous markers). Unifying concepts of major contributors to autistic symptoms include increased vulnerability to oxidative stress, immune glutamatergic dysfunction, and pineal gland malfunction. Data reviewed in this manuscript include only those that were statistically significant (p < 0.05) when compared to age- and sex-matched neurotypical control individuals. The number of subjects across the studies varied greatly. Without further analyses, it is not possible to say how much of a change in expression of a biomarker would be indicative of autism.
No single biomarker is considered specific for autism. Thus, in order to indicate autism, which is a spectrum of disorders, single markers will be inadequate. However, it is possible that a composite of multiple biomarker arrays will be able to distinguish the different autism spectrum disorders from each other and, with the use of bioinformatics and biostatistics, establish mutually inclusive discriminators that will objectively define autism. Changes in each autistic patient’s condition will most likely be manifested in metabolic changes when measured in the composite biomarker array. Longitudinal analysis of the array should be able to not only help to determine the extent of brain dysfunction, but will measure improvement caused by the patient-specific therapeutic administration of those markers which are lower than normal, and the use of antagonists for those markers which are greater than normal.
Acknowledgements
Robert B. Sothern, PhD, from the University of Minnesota, Erika Papp Faber; and, the reference department of the Danbury Public Library, Danbury, CT, are gratefully acknowledged here for their editorial assistance in the preparation of this manuscript and assistance in retrieval of original references.
Declaration of Interest
The author reports no declarations of interest. The author alone is responsible for the content and writing of the paper.
References
- Ahloulay, M., Déchaux, M., Hassler, C., Bouby, N. and Bankir, L. 1996. Cyclic AMP is a hepatorenal link influencing natriuresis and contributing to glucagon-induced hyperfiltration in rats. J. Clin. Invest. 98:2251–2258.
- Aldred, S., Moore, K.M., Fitzgerald, M. and Waring, R.H. 2003. Plasma amino acid levels in children with autism and their families. J. Autism Dev. Disord. 33:93–97.
- Anderson, C. 2010. Careful counting: How many people have an ASD? Available at: www.iancommunity.org/cs/understanding_research/prevalence (March 2, 2010).
- Arnold, G.L., Hyman, S.L., Mooney, R.A. and Kirby, R.S. 2003. Plasma amino acids profiles in children with autism: potential risk of nutritional deficiencies. J. Autism Dev. Disord. 33:449–454.
- Auyeung, B., Baron-Cohen, S., Ashwin, E., Knickmeyer, R., Taylor, K. and Hackett, G. 2009. Fetal testosterone and autistic traits. Br. J. Psychol. 100:1–22.
- Axelrod, J. 1974. The pineal gland: a neurochemical transducer. Science. 184:1341–1348.
- Axt, A. 1996. Autism viewed as a consequence of pineal gland malfunction. Farmakoter. Psychiat. Neuro. 8:112–134.
- Bannon, A.W., Decker, M.W., Holladay, M.W., Curzon, P., Donnelly-Roberts, D., Puttfarcken, P.S., Bitner, R.S., Diaz, A., Dickenson, A.H., Porsolt, R.D., Williams, M. and Arneric, S.P. 1998. Broad-spectrum, non-opioid analgesic activity by selective modulation of neuronal nicotinic acetylcholine receptors. Science. 279:77–81.
- Bauman, M.L. and Kemper, T.L. (Eds.) 1994. Neuroanatomic observations of the brain in autism. In: The Neurobiology of Autism. Baltimore: Johns Hopkins University Press, pp. 119–145.
- Bauman, M.L. and Kemper, T.L. 2005. Neuroanatomic observations of the brain in autism: a review and future directions. Int. J. Dev. Neurosci. 23:183–187.
- Belmonte, M.K., Cook, E.H. Jr, Anderson, G.M., Rubenstein, J.L., Greenough, W.T., Beckel-Mitchener, A., Courchesne, E., Boulanger, L.M., Powell, S.B., Levitt, P.R., Perry, E.K., Jiang, Y.H., DeLorey, T.M. and Tierney, E. 2004. Autism as a disorder of neural information processing: directions for research and targets for therapy. Mol. Psychiatry. 9:646–663.
- Binstock, T. 2001. Intra-monocyte pathogens delineate autism subgroups. Med. Hypotheses. 56:523–531.
- Blatt, G.J., Fitzgerald, C.M., Guptill, J.T., Booker, A.B., Kemper, T.L. and Bauman, M.L. 2001. Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J. Autism Dev. Disord. 31:537–543.
- Blaylock, R.L. and Strunecka, A. 2009. Immune-glutamatergic dysfunction as a central mechanism of the autism spectrum disorders. Curr. Med. Chem. 16:157–170.
- Brainard, G.C. 1978. Pineal research: the decade of transformation. J. Neural Transm. Suppl. 3–10.
- Buller, K.M. and Day, T.A. 2002. Systemic administration of interleukin-1beta activates select populations of central amygdala afferents. J. Comp. Neurol. 452:288–296.
- Burgess, N.K., Sweeten, T.L., McMahon, W.M. and Fujinami, R.S. 2006. Hyperserotoninemia and altered immunity in autism. J. Autism Dev. Disord. 36:697–704.
- Carlsson, M.L. 1998. Hypothesis: is infantile autism a hypoglutamatergic disorder? Relevance of glutamate - serotonin interactions for pharmacotherapy. J. Neural Transm. 105:525–535.
- Cass, H., Gringras, P., March, J., McKendrick, I., O’Hare, A.E., Owen, L. and Pollin, C. 2008. Absence of urinary opioid peptides in children with autism. Arch. Dis. Child. 93:745–750.
- Cazzullo, A.G., Musetti, M.C., Musetti, L., Bajo, S., Sacerdote, P. and Panerai, A. 1999. Beta-endorphin levels in peripheral blood mononuclear cells and long-term naltrexone treatment in autistic children. Eur. Neuropsychopharmacol. 9:361–366.
- Chauhan, A. and Chauhan, V. 2006. Oxidative stress in autism. Pathophysiology. 13:171–181.
- Chen, W.G., Chang, Q., Lin, Y., Meissner, A., West, A.E., Griffith, E.C., Jaenisch, R. and Greenberg, M.E. 2003. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 302:885–889.
- Cheng, L., Ge, Q., Xiao, P., Sun, B., Ke, X., Bai, Y. and Lu, Z. 2009. Association study between BDNF gene polymorphisms and autism by three-dimensional gel-based microarray. Int. J. Mol. Sci. 10:2487–2500.
- Chugani, D.C., Muzik, O., Behen, M., Rothermel, R., Janisse, J.J., Lee, J. and Chugani, H.T. 1999. Developmental changes in brain serotonin synthesis capacity in autistic and nonautistic children. Ann. Neurol. 45:287–295.
- Connolly, A.M., Chez, M., Streif, E.M., Keeling, R.M., Golumbek, P.T., Kwon, J.M., Riviello, J.J., Robinson, R.G., Neuman, R.J. and Deuel, R.M. 2006. Brain-derived neurotrophic factor and autoantibodies to neural antigens in sera of children with autistic spectrum disorders, Landau-Kleffner syndrome, and epilepsy. Biol. Psychiatry. 59:354–363.
- Corbett, B.A., Mendoza, S., Abdullah, M., Wegelin, J.A. and Levine, S. 2006. Cortisol circadian rhythms and response to stress in children with autism. Psychoneuroendocrinology. 31:59–68.
- Corbett, B.A., Mendoza, S., Wegelin, J.A., Carmean, V. and Levine, S. 2008. Variable cortisol circadian rhythms in children with autism and anticipatory stress. J. Psychiatry Neurosci. 33:227–234.
- Curin, J.M., Terzic, J., Petkovic, Z.B., Zekan, L., Terzic, I.M. and Susnjara, I.M. 2003. Lower cortisol and higher ACTH levels in individuals with autism. J. Autism Dev. Disord. 33:443–448.
- Davis, M., Rainnie, D. and Cassell, M. 1994. Neurotransmission in the rat amygdala related to fear and anxiety. Trends Neurosci. 17:208–214.
- DeLorey, T.M., Handforth, A., Anagnostaras, S.G., Homanics, G.E., Minassian, B.A., Asatourian, A., Fanselow, M.S., Delgado-Escueta, A., Ellison, G.D. and Olsen, R.W. 1998. Mice lacking the beta3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J. Neurosci. 18:8505–8514.
- DeMyer, M.K., Hingtgen, J.N. and Jackson, R.K. 1981. Infantile autism reviewed: a decade of research. Schizophr. Bull. 7:388–451.
- Denney, D.R., Frei, B.W. and Gaffney, G.R. 1996. Lymphocyte subsets and interleukin-2 receptors in autistic children. J. Autism Dev. Disord. 26:87–97.
- Elchaar, G.M., Maisch, N.M., Augusto, L.M. and Wehring, H.J. 2006. Efficacy and safety of naltrexone use in pediatric patients with autistic disorder. Ann. Pharmacother. 40:1086–1095.
- Evans, C., Dunstan, R.H., Rothkirch, T., Roberts, T.K., Reichelt, K.L., Cosford, R., Deed, G., Ellis, L.B. and Sparkes, D.L. 2008. Altered amino acid excretion in children with autism. Nutr. Neurosci. 11:9–17.
- Feldman, H.M., Kolmen, B.K. and Gonzaga, A.M. 1999. Naltrexone and communication skills in young children with autism. J. Am. Acad. Child Adolesc. Psychiatry. 38:587–593.
- Filipek, P.A., Juranek, J., Nguyen, M.T., Cummings, C. and Gargus, J.J. 2004. Relative carnitine deficiency in autism. J. Autism Dev. Disord. 34:615–623.
- Fuchs, D., Weiss, G. and Wachter, H. 1993. Neopterin, biochemistry and clinical use as a marker for cellular immune reactions. Int. Arch. Allergy Immunol. 101:1–6.
- Galli-Carminati, G., Deriaz, N. and Bertschy, G. 2009. Melatonin in treatment of chronic sleep disorders in adults with autism: a retrospective study. Swiss Med. Wkly. 139:293–296.
- Geier, D.A. and Geier, M.R. 2006a. A prospective assessment of porphyrins in autistic disorders: a potential marker for heavy metal exposure. Neurotox. Res. 10:57–64.
- Geier, D.A. and Geier, M.R. 2006b. A clinical and laboratory evaluation of methionine cycle-transsulfuration and androgen pathway markers in children with autistic disorders. Horm. Res. 66:182–188.
- Geier, D.A., Kern, J.K., Garver, C.R., Adams, J.B., Audhya, T., Nataf, R. and Geier, M.R. 2009. Biomarkers of environmental toxicity and susceptibility in autism. J. Neurol. Sci. 280:101–108.
- Giannotti, F., Cortesi, F., Cerquiglini, A. and Bernabei, P. 2006. An open-label study of controlled-release melatonin in treatment of sleep disorders in children with autism. J. Autism Dev. Disord. 36:741–752.
- Gillberg, C. 1995. Endogenous opioids and opiate antagonists in autism: brief review of empirical findings and implications for clinicians. Dev. Med. Child Neurol. 37:239–245.
- Glickman, G. 2010. Circadian rhythms and sleep in children with autism. Neurosci. Biobehav. Rev. 34:755–768.
- Grigorenko, E.L., Han, S.S., Yrigollen, C.M., Leng, L., Mizue, Y., Anderson, G.M., Mulder, E.J., de Bildt, A., Minderaa, R.B., Volkmar, F.R., Chang, J.T. and Bucala, R. 2008. Macrophage migration inhibitory factor and autism spectrum disorders. Pediatrics. 122:e438–e445.
- Hardan, A.Y. and Handen, B.L. 2002. A retrospective open trial of adjunctive donepezil in children and adolescents with autistic disorder. J. Child Adolesc. Psychopharmacol. 12:237–241.
- Hare, D.J., Jones, S. and Evershed, K. 2006. A comparative study of circadian rhythm functioning and sleep in people with Asperger syndrome. Autism. 10:565–575.
- Hashimoto, K., Iwata, Y., Nakamura, K., Tsujii, M., Tsuchiya, K.J., Sekine, Y., Suzuki, K., Minabe, Y., Takei, N., Iyo, M. and Mori, N. 2006. Reduced serum levels of brain-derived neurotrophic factor in adult male patients with autism. Prog. Neuropsychopharmacol. Biol. Psychiatry. 30:1529–1531.
- Hohmann, C.F. and Berger-Sweeney, J. 1998. Cholinergic regulation of cortical development and plasticity. New twists to an old story. Perspect. Dev. Neurobiol. 5:401–425.
- Hollander, E., DelGiudice-Asch, G., Simon, L., Schmeidler, J., Cartwright, C., DeCaria, C.M., Kwon, J., Cunningham-Rundles, C., Chapman, F. and Zabriskie, J.B. 1999. B lymphocyte antigen D8/17 and repetitive behaviors in autism. Am. J. Psychiatry. 156:317–320.
- Holmes, C., El-Okl, M., Williams, A.L., Cunningham, C., Wilcockson, D. and Perry, V.H. 2003. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatr. 74:788–789.
- Horvath, K., Papadimitriou, J.C., Rabsztyn, A., Drachenberg, C. and Tildon, J.T. 1999. Gastrointestinal abnormalities in children with autistic disorder. J. Pediatr. 135:559–563.
- Horvath, K. and Perman, J.A. 2002. Autistic disorder and gastrointestinal disease. Curr. Opin. Pediatr. 14:583–587.
- Hoshino, Y., Kumashiro, H., Yashima, Y., Kaneko, M., Numata, Y., Oshima, N. and Watanabe, A. 1980. Plasma cyclic AMP level in psychiatric diseases of childhood. Folia Psychiatr. Neurol. Jpn. 34:9–16.
- Hoshino, Y., Kumashiro, H., Yashima, Y., Tachibana, R., Watanabe, M. and Furukawa, H. 1982. Early symptoms of autistic children and its diagnostic significance. Folia Psychiatr. Neurol. Jpn. 36:367–374.
- Hunter, L.C., O’Hare, A., Herron, W.J., Fisher, L.A. and Jones, G.E. 2003. Opioid peptides and dipeptidyl peptidase in autism. Dev. Med. Child Neurol. 45:121–128.
- Jamain, S., Betancur, C., Quach, H., Philippe, A., Fellous, M., Giros, B., Gillberg, C., Leboyer, M. and Bourgeron, T. Paris Autism Research International Sibpair (PARIS) Study. 2002. Linkage and association of the glutamate receptor 6 gene with autism. Mol. Psychiatry. 7:302–310.
- James, S.J., Cutler, P., Melnyk, S., Jernigan, S., Janak, L., Gaylor, D.W. and Neubrander, J.A. 2004. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 80:1611–1617.
- Jepson B. (Ed.) 2007a. Changing the Course of Autism. Boulder, CO: Sentient Publications, pp. 176–180.
- Jepson B. (Ed.) 2007b. Changing the Course of Autism. Boulder, CO: Sentient Publications, pp. 225.
- Jepson B. (Ed.) 2007c. Changing the Course of Autism. Boulder, CO: Sentient Publications, pp. 226.
- Jepson B. (Ed.) 2007d. Changing the Course of Autism. Boulder, CO: Sentient Publications, pp. 227.
- Jyonouchi, H., Sun, S. and Le, H. 2001. Proinflammatory and regulatory cytokine production associated with innate and adaptive immune responses in children with autism spectrum disorders and developmental regression. J. Neuroimmunol. 120:170–179.
- Kaplan, D.R. and Miller, F.D. 2000. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 10:381–391.
- Kaufman, S. 1986. The metabolic role of tetrahydrobiopterin. In: Chemistry and Biology of Pteridines (Cooper, B.A. and Whitehead, V.A., Eds.), Berlin: Walter de Gruyter, pp. 185–200.
- Kelley, D.J., Bhattacharyya, A., Lahvis, G.P., Yin, J.C., Malter, J. and Davidson, R.J. 2008. The cyclic AMP phenotype of Fragile X and autism. Neurosci. Biobehav. Rev. 32:1533–1543.
- Kelley, R.I. and Hennekam, R.C. 2000. Smith-Lemli-Opitz syndrome. In: The Metabolic and Molecular Basics of Inherited Disease, 8th Edit. (Scriver, C.R., Beaudet, A.L., Sly, W.S. and Valle, D., Eds.), New York: McGraw-Hill, pp. 6183–6201.
- Kemper, T.L. and Bauman, M. 1998. Neuropathology of infantile autism. J. Neuropathol. Exp. Neurol. 57:645–652.
- Kitay, J.L. and Altschule, M.P. (Eds.) 1954. The Pineal Gland. Cambridge, MA: Harvard Univ. Press, pp. 72–74.
- Klauck, S.M., Poustka, F., Benner, A., Lesch, K.P. and Poustka, A. 1997. Serotonin transporter (5-HTT) gene variants associated with autism? Hum. Mol. Genet. 6:2233–2238.
- Knickmeyer, R., Baron-Cohen, S., Raggatt, P. and Taylor, K. 2005. Foetal testosterone, social relationships, and restricted interests in children. J. Child Psychol. Psychiatry. 46:198–210.
- Köhler, M., Assmann, B., Bräutigam, C., Storm, W., Marie, S., Vincent, M.F., Van den Berghe, G., Simmonds, H.A. and Hoffmann, G.F. 1999. Adenylosuccinase deficiency: possibly underdiagnosed encephalopathy with variable clinical features. Eur. J. Paediatr. Neurol. 3:3–6.
- Kolvin, I. 1971. Studies in the childhood psychoses. I. Diagnostic criteria and classification. Br. J. Psychiatry. 118:381–384.
- Koukkari, W.L. and Sothern, R.B. (Eds.) 2006. Introducing Biological Rhythms. New York: Springer, pp. 1–18.
- Kulman, G., Lissoni, P., Rovelli, F., Roselli, M.G., Brivio, F. and Sequeri, P. 2000. Evidence of pineal endocrine hypofunction in autistic children. Neuro Endocrinol. Lett. 21:31–34.
- Kunkel, S.L., Ward, P.A., Caporale, L.H. and Vogel, C.W. 1985. The Complement System In: Immunology III (Bellanti, J. A., Ed.). Philadelphia: W.B. Saunders Co., pp. 106–116.
- Laurie, D.J., Wisden, W. and Seeburg, P.H. 1992. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J. Neurosci. 12:4151–4172.
- Leboyer, M., Philippe, A., Bouvard, M., Guilloud-Bataille, M., Bondoux, D., Tabuteau, F., Feingold, J., Mouren-Simeoni, M.C. and Launay, J.M. 1999. Whole blood serotonin and plasma beta-endorphin in autistic probands and their first-degree relatives. Biol. Psychiatry. 45:158–163.
- Lee, M., Martin-Ruiz, C., Graham, A., Court, J., Jaros, E., Perry, R., Iversen, P., Bauman, M. and Perry, E. 2002. Nicotinic receptor abnormalities in the cerebellar cortex in autism. Brain. 125:1483–1495.
- Lerner, A.B., Case, J.D., Takahashi, Y., Lee, T.H. and Mori, W. 1958. Isolation of melatonin, the pineal gland factor that lightens melanocytes. J. Am. Chem. Soc. 80:2587.
- Lerner, A.B., Case, J.D. and Takahashi, Y. 1960. Isolation of melatonin and 5-methoxyindole-3-acetic acid from bovine pineal glands. J. Biol. Chem. 235:1992–1997.
- Lord, C., Rutter, M. and Le Couteur, A. 1994. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 24:659–685.
- Luboshitzky, R., Wagner, O., Lavi, S., Herer, P. and Lavie, P. 1996. Decreased nocturnal melatonin secretion in patients with Klinefelter’s syndrome. Clin. Endocrinol. (Oxf). 45:749–754.
- Luboshitzky, R., Wagner, O., Lavi, S., Herer, P. and Lavie, P. 1997. Abnormal melatonin secretion in hypogonadal men: the effect of testosterone treatment. Clin. Endocrinol. (Oxf). 47:463–469.
- Lucki, I. 1998. The spectrum of behaviors influenced by serotonin. Biol. Psychiatry. 44:151–162.
- Malhotra, S., Sawhney, G. and Pandhi, P. 2004. The therapeutic potential of melatonin: a review of the science. MedGenMed. 6:46.
- Malow, B.A., Marzec, M.L., McGrew, S.G., Wang, L., Henderson, L.M. and Stone, W.L. 2006. Characterizing sleep in children with autism spectrum disorders: a multidimensional approach. Sleep. 29:1563–1571.
- Marinovic-Curin, J., Marinovic-Terzic, I., Bujas-Petkovic, Z., Zekan, L., Skrabic, V., Dogas, Z. and Terzic, J. 2008. Slower cortisol response during ACTH stimulation test in autistic children. Eur. Child Adolesc. Psychiatry. 17:39–43.
- Martineau, J., Barthélémy, C., Jouve, J., Muh, J.P. and Lelord, G. 1992. Monoamines (serotonin and catecholamines) and their derivatives in infantile autism: age-related changes and drug effects. Dev. Med. Child Neurol. 34:593–603.
- Martin-Ruiz, C.M., Lee, M., Perry, R.H., Baumann, M., Court, J.A. and Perry, E.K. 2004. Molecular analysis of nicotinic receptor expression in autism. Brain Res. Mol. Brain Res. 123:81–90.
- Megson, M.N. 2000. Is autism a G-alpha protein defect reversible with natural vitamin A? Med. Hypotheses. 54:979–983.
- Mehl-Madrona, L. 2000. Autism: An overview. www.healing-arts.org/children/autism-overview.htm.
- Melke, J., Goubran Botros, H., Chaste, P., Betancur, C., Nygren, G., Anckarsäter, H., Rastam, M., Ståhlberg, O., Gillberg, I.C., Delorme, R., Chabane, N., Mouren-Simeoni, M.C., Fauchereau, F., Durand, C.M., Chevalier, F., Drouot, X., Collet, C., Launay, J.M., Leboyer, M., Gillberg, C. and Bourgeron, T. 2008. Abnormal melatonin synthesis in autism spectrum disorders. Mol. Psychiatry. 13:90–98.
- Messahel, S., Pheasant, A.E., Pall, H., Ahmed-Choudhury, J., Sungum-Paliwal, R.S. and Vostanis, P. 1998. Urinary levels of neopterin and biopterin in autism. Neurosci. Lett. 241:17–20.
- Miyashiro, K. and Eberwine, J. 2004. Fragile X syndrome: (What’s) lost in translation? Proc. Natl. Acad. Sci. U.S.A. 101:17329–17330.
- Moreno, H., Borjas, L., Arrieta, A., Sáez, L., Prassad, A., Estévez, J. and Bonilla, E. 1992. [Clinical heterogeneity of the autistic syndrome: a study of 60 families]. Invest. Clin. 33:13–31.
- Mössner, R. and Lesch, K.P. 1998. Role of serotonin in the immune system and in neuroimmune interactions. Brain Behav. Immun. 12:249–271.
- Muhle, R., Trentacoste, S.V. and Rapin, I. 2004. The genetics of autism. Pediatrics. 113:e472–e486.
- Nataf, R., Skorupka, C., Amet, L., Lam, A., Springbett, A. and Lathe, R. 2006. Porphyrinuria in childhood autistic disorder: implications for environmental toxicity. Toxicol. Appl. Pharmacol. 214:99–108.
- Nelson, K.B., Grether, J.K., Croen, L.A., Dambrosia, J.M., Dickens, B.F., Jelliffe, L.L., Hansen, R.L. and Phillips, T.M. 2001. Neuropeptides and neurotrophins in neonatal blood of children with autism or mental retardation. Ann. Neurol. 49:597–606.
- Nir, I., Meir, D., Zilber, N., Knobler, H., Hadjez, J. and Lerner, Y. 1995. Brief report: circadian melatonin, thyroid-stimulating hormone, prolactin, and cortisol levels in serum of young adults with autism. J. Autism Dev. Disord. 25:641–654.
- Norman, A.W. and Litwach, G. (Eds.) 1987. Hormones. New York: Academic Press, Inc., pp. 711.
- Ornitz, E.M., Ritvo, E.R. and Walter, R.D. 1965. Dreaming sleep in autistic and schizophrenic children. Am. J. Psychiatry. 122:419–424.
- Ornitz, E.M. 1973. Childhood autism. A review of the clinical and experimental literature. Calif. Med. 118:21–47.
- Panksepp, J. 1979. A neurochemical theory of autism. Trends Neurosci. 2:174–177.
- Pennell, P.B., Burdette, D.E., Ross, D.A., Henry, T.R., Albin, R.L., Sackellares, J.C. and Frey, K.A. 1999. Muscarinic receptor loss and preservation of presynaptic cholinergic terminals in hippocampal sclerosis. Epilepsia. 40:38–46.
- Perry, E.K., Lee, M.L., Martin-Ruiz, C.M., Court, J.A., Volsen, S.G., Merrit, J., Folly, E., Iversen, P.E., Bauman, M.L., Perry, R.H. and Wenk, G.L. 2001. Cholinergic activity in autism: abnormalities in the cerebral cortex and basal forebrain. Am. J. Psychiatry. 158:1058–1066.
- Plioplys, A.V., Greaves, A., Kazemi, K. and Silverman, E. 1994. Lymphocyte function in autism and Rett syndrome. Neuropsychobiology. 29:12–16.
- Polleux, F. and Lauder, J.M. 2004. Toward a developmental neurobiology of autism. Ment. Retard. Dev. Disabil. Res. Rev. 10:303–317.
- Posey, D.J., Stigler, K.A., Erickson, C.A. and McDougle, C.J. 2008. Antipsychotics in the treatment of autism. J. Clin. Invest. 118:6–14.
- Ratajczak, H.V. 2010. Theoretical aspects.of autism: Causes – A review. J. Immunotoxicol. (submitted for publication).
- Reichelt, K.L. and Liu, Y. 1997. Presentation at Defeat Autism Now! Meeting, San Diego, CA. Sept 19–21.
- Reichelt, K.L. and Knivsberg, A.M. 2003. Can the pathophysiology of autism be explained by the nature of the discovered urine peptides? Nutr. Neurosci. 6:19–28.
- Reiter, R. J., and Robinson, J. (Eds.) 1995a. Melatonin. New York: Bantam Books, pp. 22–23.
- Reiter, R. J., and Robinson, J. (Eds.) 1995b. Melatonin. New York: Bantam Books, p. 3.
- Reiter, R. J., and Robinson, J. (Eds.) 1995c. Melatonin. New York: Bantam Books, pp. 3–12.
- Reiter, R.J., Tan, D.X. and Fuentes-Broto, L. 2010. Melatonin: a multitasking molecule. Prog. Brain Res. 181:127–151.
- Renzoni, E., Beltrami, V., Sestini, P., Pompella, A., Menchetti, G. and Zappella, M. 1995. Brief report: allergological evaluation of children with autism. J. Autism Dev. Disord. 25:327–333.
- Riddle, M.A., Bernstein, G.A., Cook, E.H., Leonard, H.L., March, J.S. and Swanson, J.M. 1999. Anxiolytics, adrenergic agents, and naltrexone. J. Am. Acad. Child Adolesc. Psychiatry. 38:546–556.
- Rolf, L.H., Haarmann, F.Y., Grotemeyer, K.H. and Kehrer, H. 1993. Serotonin and amino acid content in platelets of autistic children. Acta Psychiatr. Scand. 87:312–316.
- Rubenstein, J.L. and Merzenich, M.M. 2003. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2:255–267.
- Sabbagh, M.N., Reid, R.T., Corey-Bloom, J., Rao, T.S., Hansen, L.A., Alford, M., Masliah, E., Adem, A., Lloyd, G.K. and Thal, L.J. 1998. Correlation of nicotinic binding with neurochemical markers in Alzheimer’s disease. J. Neural Transm. 105:709–717.
- Sanchez de la Pena, S. 1993. The feed-sideward of cephalo-adrenal immune interactions. Chronobiologia 20:1–52.
- Selmaoui, B. and Touitou, Y. 2003. Reproducibility of the circadian rhythms of serum cortisol and melatonin in healthy subjects: a study of three different 24-h cycles over six weeks. Life Sci. 73:3339–3349.
- Serajee, F.J., Zhong, H., Nabi, R. and Huq, A.H. 2003. The metabotropic glutamate receptor 8 gene at 7q31: partial duplication and possible association with autism. J. Med. Genet. 40:e42.
- Shanahan, M.R., Venturini, A.J., Daiss, J.L. and Friedman, A.E. 2000. Peptide diagnostic markers for human disorders. European Patent Application. Ep 0 969 015 A2. 05 Jan.
- Shattock, P. and Whiteley, P. 2002. Biochemical aspects in autism spectrum disorders: updating the opioid-excess theory and presenting new opportunities for biomedical intervention. Expert Opin. Ther. Targets. 6:175–183.
- Singh, V.K., Warren, R.P., Odell, J.D. and Cole, P. 1991. Changes of soluble interleukin-2, interleukin-2 receptor, T8 antigen, and interleukin-1 in the serum of autistic children. Clin. Immunol. Immunopathol. 61:448–455.
- Singh, V.K., Warren, R.P., Odell, J.D., Warren, W.L. and Cole, P. 1993. Antibodies to myelin basic protein in children with autistic behavior. Brain Behav. Immun. 7:97–103.
- Singh, V.K. 1996. Plasma increase of interleukin-12 and interferon-gamma. Pathological significance in autism. J. Neuroimmunol. 66:143–145.
- Singh, V.K., Warren, R., Averett, R. and Ghaziuddin, M. 1997. Circulating autoantibodies to neuronal and glial filament proteins in autism. Pediatr. Neurol. 17:88–90.
- Slauson, D.O., Walker, C., Kristensen, F., Wang, Y. and de Weck, A.L. 1984. Mechanisms of serotonin-induced lymphocyte proliferation inhibition. Cell. Immunol. 84:240–252.
- Stefulj, J., Cicin-Sain, L., Schauenstein, K. and Jernej, B. 2001. Serotonin and immune response: effect of the amine on in vitro proliferation of rat lymphocytes. Neuroimmunomodulation. 9:103–108.
- Stubbs, E.G. and Crawford, M.L. 1977. Depressed lymphocyte responsiveness in autistic children. J. Autism Child. Schizophr. 7:49–55.
- Sweeten, T.L., Posey, D.J. and McDougle, C.J. 2003. High blood monocyte counts and neopterin levels in children with autistic disorder. Am. J. Psychiatry. 160:1691–1693.
- Tierney, E., Nwokoro, N.A., Porter, F.D., Freund, L.S., Ghuman, J.K. and Kelley, R.I. 2001. Behavior phenotype in the RSH/Smith-Lemli-Opitz syndrome. Am. J. Med. Genet. 98:191–200.
- Tint, G.S., Irons, M., Elias, E.R., Batta, A.K., Frieden, R., Chen, T.S. and Salen, G. 1994. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N. Engl. J. Med. 330:107–113.
- Tordjman, S., Anderson, G.M., McBride, P.A., Hertzig, M.E., Snow, M.E., Hall, L.M., Ferrari, P. and Cohen, D.J. 1995. Plasma androgens in autism. J. Autism Dev. Disord. 25:295–304.
- Tordjman, S., Anderson, G.M., McBride, P.A., Hertzig, M.E., Snow, M.E., Hall, L.M., Thompson, S.M., Ferrari, P. and Cohen, D.J. 1997. Plasma beta-endorphin, adrenocorticotropin hormone, and cortisol in autism. J. Child Psychol. Psychiatry. 38:705–715.
- Tordjman, S., Antoine, C., Cohen, D.J., Gauvain-Piquard, A., Carlier, M., Roubertoux, P. and Ferrari, P. 1999. [Study of the relationships between self-injurious behavior and pain reactivity in infantile autism]. Encephale. 25:122–134.
- Tordjman, S., Anderson, G.M., Pichard, N., Charbuy, H. and Touitou, Y. 2005. Nocturnal excretion of 6-sulphatoxymelatonin in children and adolescents with autistic disorder. Biol. Psychiatry. 57:134–138.
- Tordjman, S., Anderson, G.M., Botbol, M., Brailly-Tabard, S., Perez-Diaz, F., Graignic, R., Carlier, M., Schmit, G., Rolland, A.C., Bonnot, O., Trabado, S., Roubertoux, P. and Bronsard, G. 2009. Pain reactivity and plasma β-endorphin in children and adolescents with autistic disorder. PLoS ONE 4(8):e5289, doi:10.1371/journal.pone.0005289.
- Vancassel, S., Durand, G., Barthélémy, C., Lejeune, B., Martineau, J., Guilloteau, D., Andrès, C. and Chalon, S. 2001. Plasma fatty acid levels in autistic children. Prostaglandins Leukot. Essent. Fatty Acids 65:1–7.
- Vargas, D.L., Nascimbene, C., Krishnan, C., Zimmerman, A.W. and Pardo, C.A. 2005. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 57:67–81.
- Vojdani, A., Bazargan, M., Vojdani, E., Samadi, J., Nourian, A.A., Eghbalieh, N. and Cooper, E.L. 2004a. Heat shock protein and gliadin peptide promote development of peptidase antibodies in children with autism and patients with autoimmune disease. Clin. Diagn. Lab. Immunol. 11:515–524.
- Vojdani, A., O’Bryan, T., Green, J.A., Mccandless, J., Woeller, K.N., Vojdani, E., Nourian, A.A. and Cooper, E.L. 2004b. Immune response to dietary proteins, gliadin and cerebellar peptides in children with autism. Nutr. Neurosci. 7:151–161.
- Volkmar, F.R., Lord, C., Bailey, A., Schultz, R.T. and Klin, A. 2004. Autism and pervasive developmental disorders. J. Child Psychol. Psychiatry 45:135–170.
- Volkmar, F. R., and Klin, A. 2005. Issues in the classification of autism and related conditions In: Handbook of Autism and Pervasive Developmental Disorders, 3rd Edition. (Volkmar, F.R., Paul, R., Klin, A., Cohen, D., Eds.), Hoboken, NJ: John Wiley & Sons, Inc., pp. 5–14.
- Wakefield, A.J., Murch, S.H., Anthony, A., Linnell, J., Casson, D.M., Malik, M., Berelowitz, M., Dhillon, A.P., Thomson, M.A., Harvey, P., Valentine, A., Davies, S.E. and Walker-Smith, J.A. 1998. Ileal-lymphoid-nodular hyperplasia, non-specific colitis, and pervasive developmental disorder in children. Lancet 351:637–641.
- Ward, N.L. and Hagg, T. 2000. BDNF is needed for postnatal maturation of basal forebrain and neostriatum cholinergic neurons in vivo. Exp. Neurol. 162:297–310.
- Waring, R. H., Ngong, J. M., Klovarza, L., Green, S., and Sharp, H. 1997. Biochemical parameters in autistic children. Dev. Brain Dysfunc. 10:40–43.
- Waring, R. H., and Klovrza, L. V. 2000. Sulfur metabolism in autism. J. Nutr. Environ. Med. 10:25–32.
- Warren, R.P., Margaretten, N.C., Pace, N.C. and Foster, A. 1986. Immune abnormalities in patients with autism. J. Autism Dev. Disord. 16:189–197.
- Warren, R.P., Foster, A. and Margaretten, N.C. 1987. Reduced natural killer cell activity in autism. J. Am. Acad. Child Adolesc. Psychiatry 26:333–335.
- Warren, R.P., Yonk, L.J., Burger, R.A., Cole, P., Odell, J.D., Warren, W.L., White, E. and Singh, V.K. 1990. Deficiency of suppressor-inducer (CD4+CD45RA+) T-cells in autism. Immunol. Invest. 19:245–251.
- Warren, R.P., Singh, V.K., Cole, P., Odell, J.D., Pingree, C.B., Warren, W.L. and White, E. 1991. Increased frequency of the null allele at the complement C4b locus in autism. Clin. Exp. Immunol. 83:438–440.
- Warren, R.P., Burger, R.A., Odell, D., Torres, A.R. and Warren, W.L. 1994. Decreased plasma concentrations of the C4B complement protein in autism. Arch. Pediatr. Adolesc. Med. 148:180–183.
- Warren, R.P., Yonk, J., Burger, R.W., Odell, D. and Warren, W.L. 1995. DR-positive T-cells in autism: Association with decreased plasma levels of the complement C4B protein. Neuropsychobiology 31:53–57.
- Warren, R.P., Singh, V.K., Averett, R.E., Odell, J.D., Maciulis, A., Burger, R.A., Daniels, W.W. and Warren, W.L. 1996. Immunogenetic studies in autism and related disorders. Mol. Chem. Neuropathol. 28:77–81.
- Warren, R.P., Odell, J.D., Warren, W.L., Burger, R.A., Maciulis, A., Daniels, W.W. and Torres, A.R. 1997. Brief report: immunoglobulin A deficiency in a subset of autistic subjects. J. Autism Dev. Disord. 27:187–192.
- Weizman, A., Weizman, R., Szekely, G.A., Wijsenbeek, H. and Livni, E. 1982. Abnormal immune response to brain tissue antigen in the syndrome of autism. Am. J. Psychiatry. 139:1462–1465.
- Werner, E.R., Werner-Felmayer, G., Fuchs, D., Hausen, A., Reibnegger, G. and Wachter, H. 1989. Parallel induction of tetrahydrobiopterin biosynthesis and indoleamine 2,3-dioxygenase activity in human cells and cell lines by interferon-gamma. Biochem. J. 262:861–866.
- Whitaker-Azmitia, P.M. 2001. Serotonin and brain development: Role in human developmental diseases. Brain Res. Bull. 56:479–485.
- Wikipedia. 2010. Available at http://en.wikipedia.org/’wiki/Interior_olivary_nucleus.
- Willemsen-Swinkels, S.H., Buitelaar, J.K., Nijhof, G.J. and van England, H. 1995. Failure of naltrexone hydrochloride to reduce self-injurious and autistic behavior in mentally retarded adults. Double-blind placebo-controlled studies. Arch. Gen. Psychiatry 52:766–773.
- Wing, L. 2005. Problems of categorical classification systems. In: Handbook of Autism and Pervasive Developmental Disorders. Volume 1. 3rd Ed. (Volkmar, F. R., Paul, R., Klin, A., and Cohen, D., Eds.) Hoboken, NJ: Wiley, pp. 583–605.
- Wirojanan, J., Jacquemont, S., Diaz, R., Bacalman, S., Anders, T.F., Hagerman, R.J. and Goodlin-Jones, B.L. 2009. The efficacy of melatonin for sleep problems in children with autism, Fragile X syndrome, or autism and Fragile X syndrome. J. Clin. Sleep Med. 5:145–150.
- Yonk, L.J., Warren, R.P., Burger, R.A., Cole, P., Odell, J.D., Warren, W.L., White, E. and Singh, V.K. 1990. CD4+ helper T-cell depression in autism. Immunol. Lett. 25:341–345.
- Young, M.R. and Matthews, J.P. 1995. Serotonin regulation of T-cell subpopulations and of macrophage accessory function. Immunology 84:148–152.