Abstract
Propofol is an intravenous anesthetic that is widely used for anesthesia and sedation. Dendritic cells (DC) are one of the crucial immune cells that bridge innate and adaptive immunity, in which DC process antigens during innate immune responses to present them to naïve T-cells, leading to an establishment of adaptive immunity. Prostaglandin (PG)-E2 may be secreted by DC into the microenvironment, considerably influencing DC phenotype and function, and thus determining the fate of adaptive immunity. Since propofol suppresses PGE2 production in murine macrophages, the primary purpose of the present study was to determine whether propofol also suppresses PGE2 production in DC. Assuming a positive finding of such suppression, we tested whether this also leads to alterations of interleukin (IL)-12 and IL-10 production and DC surface marker expression, both of which can be modulated by PGE2. In bone marrow-derived DC, propofol significantly suppressed the PGE2 production after lipopolysaccharide stimulation. Cyclo-oxygenase (COX) protein expression and arachidonic acid release were unaffected, while COX enzyme activity was significantly inhibited by propofol. The propofol-induced COX inhibition did not lead to the increased production of cysteinyl leukotrienes and leukotriene-B4. Endogenous COX inhibition with propofol, as well as with the selective COX-2 inhibitor, NS-398, did not affect IL-12 and IL-10 production from DC. The surface expression of I-Ab and CD40 on DC was not changed, while that of CD86 slightly increased, with both propofol and NS-398; expression of CD80 was not affected with propofol, but increased slightly with NS-398. Finally, endogenous COX inhibition with either propofol or NS-398 did not significantly affect the ability of DC to induce allogeneic T-cell proliferation. It is concluded that the intravenous anesthetic propofol suppresses COX enzyme activity in DC, with no consequences with respect to IL-12/IL-10 production and allogeneic T-cell proliferation, while minimal consequences were observed in surface molecule expression.
Introduction
Dendritic cells (DC) are the only cells that can activate naïve T-cells (Sompayrac, Citation2003; Mak, Citation2006). Immature DC capture and process antigens, and present the processed antigen peptides on the major histocompatibility complex (MHC) molecules (Sompayrac, Citation2003; Mak, Citation2006). During this process, DC may also sense danger signals and produce cytokines and prostanoids in response, both of which can modify the DC microenvironment (Sompayrac, Citation2003; Mak, Citation2006). The microenvironment of DC may greatly influence the phenotype and function of DC, and thus the characteristics of subsequent adaptive immunity.
Dendritic cells are able to produce prostanoids (Norgauer et al., Citation2003; Fogel-Petrovic et al., Citation2004) from arachidonic acid (AA) by the action of cyclo-oxygenase (COX) enzymes (Needleman et al., Citation1986). COX enzymes are primarily classified as COX-1 or COX-2. COX-1 is constitutively expressed in almost all tissues, while COX-2 is usually not expressed in the resting state and is induced by various kinds of stimulation in certain cell types (Otto and Smith, Citation1995). Among prostanoids, DC predominantly produce prostaglandin (PG)-E2 and thromboxane (TX)-A2 (Gualde and Harizi, Citation2004). PGE2 is considered an immunomodulatory prostanoid, and can act on DC to stimulate production of interleukin (IL)-10 (Gualde and Harizi, Citation2004) and inhibit IL-12 production (Gualde and Harizi, Citation2004; Sa-Nunes et al., Citation2007; Shiraishi et al., Citation2008), creating a distinct DC microenvironment.
Propofol (2,6-di-isopropylphenol) is an intravenous general anesthetic widely used in the operating room for general anesthesia and in intensive care units for sedation (Vanlersberghe and Camu, Citation2008). Propofol has been reported to suppress PGE2 production from murine macrophages (Inada et al., Citation2010). Because DC phenotypically resemble macrophages, our primary purpose was to determine whether the inhibitory effect of propofol on prostanoid production observed in macrophages was also observed in DC. We also tested whether the phenotypical and functional changes that may be related to PGE2 were observed after the stimulation of DC by lipopolysaccharide (LPS) in the presence of propofol. We believe that it is important to elucidate the effect of propofol on DC because propofol is widely used to anesthetize patients undergoing surgery involving tumor resection and organ transplantation, and DC play a central role in tumor immunity as well as transplantation immunity.
Materials and methods
Reagents
The following chemical reagents were purchased from Sigma Aldrich (St. Louis, MO): propofol (2,6-di-isopropyl), LPS (Escherichia coli type 055:B5), 3-[4,5-dimethyl- thiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), A23187, and arachidonic acid. NS-398 was obtained from Cayman Chemical (Ann Arbor, MI). A non-anesthetic propofol analog, 2,6-di-tert-butylphenol, was obtained from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). Recombinant murine granulocyte macrophage colony stimulating factor (GM-CSF) and tumor necrosis factor (TNF)-α were obtained from Peprotech (London, UK). Tritiated arachidonic acid ([3H]-AA) was purchased from PerkinElmer (Yokohama, Japan).
Animals
Male C57BL/6 and BALB/c mice (8–9 weeks-of-age) were purchased from CLEA (Osaka, Japan), housed in an animal facility maintained at 20–26°C with 30–70% relative humidity and with 12-h light/dark cycles, and used according to institutional guidelines. All mice were provided access to standard rodent chow and autoclaved water ad libitum. This study was approved by the Institutional Animal Care and Use Committee of the Kansai Medical University.
Generation and culture of bone marrow-derived DC
Bone marrow-derived DC were generated according to the method of Lutz et al. (Citation1999), as previously described. Briefly, bone marrow cells were collected from the femurs and tibiae of male C57BL/6 mice. These cells (2 × 105/ml, 10 ml per dish) were cultured in 100 mm bacteriological petri dishes (Falcon, No. 1029, Beckton Dickinson, Heidelberg, Germany) in RPMI 1640 medium (Sigma) supplemented with 10% fetal calf serum (FCS) (endotoxin < 10.0 EU/ml) (HyClone, Logan, UT), 50 U penicillin/ml, 50 µg streptomycin/ml, and 55 µM 2-mercaptoethanol (Gibco, Grand Island, NY) (RPMI complete medium) containing 20 ng/ml GM-CSF.
On day 3 of culture, another 10 ml RPMI complete medium containing GM-CSF was added. On days 6 and 8, one-half of the culture medium was collected and centrifuged; the cell pellet was re-suspended in 10 ml fresh RPMI complete medium containing GM-CSF and added back to the cultures. On day 10, floating cells were collected, and CD11c+ DC were purified by magnetic sorting using anti-CD11c antibody-coated magnetic beads (Milteny Biotec, Bergisch Gladbach, Germany). The purity of the sorted CD11c+ cells was > 97%, as determined by flow cytometry.
Cells were then cultured in RPMI complete medium in flat-bottom 96-well plates (105 cells/200 µl/well) with or without LPS (1 µg/ml) in the presence or absence of various concentrations of propofol (0–30 µM) for assay-specific periods of time at 37°C in a humidified chamber containing 5% CO2. DMSO (final concentration < 0.1%) was used when propofol was absent. Cell-free supernatants were collected at dedicated timepoints (see specific assays below) and stored at −80°C before being analyzed. Propofol concentrations of 7.5, 15, and 30 µM may correspond to plasma concentrations achieved during moderate sedation, deep sedation, and anesthesia, respectively, in humans (Rigouzzo et al., Citation2008).
PGE2, TXB2, cysteinyl leukotrienes, leukotriene-B4, IL-12 (p70), and IL-10 measurements
PGE2, TXB2, cysteinyl leukotrienes (cysLT), and leukotriene-B4 (LTB4) concentrations were measured with an enzyme immunoassay (Cayman Chemical). This assay is based on the competition between PGE2 (or TXB2, cysLT, LTB4) and a PGE2 (or TXB2, cysLT, LTB4)-acetyl-cholinesterase conjugate for a limited amount of anti-PGE2 (or -TXB2, -cysLT, -LTB4) antibody. TXB2 was measured instead of TXA2 because TXA2 is rapidly hydrolyzed non-enzymatically to form TXB2. Concentrations of IL-12 (p70) and IL-10 were measured with enzyme-linked immunosorbent assays (ELISA) (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
Cell viability assay
Mitochondrial dehydrogenase changes MTT into water-insoluble formazan. The amount of formazan produced indicates mitochondrial enzyme activity, and thus cell viability. After DC maturation with LPS, supernatants were exchanged for 0.5 mg/ml MTT solution followed by incubation at 37°C for 4 h. Next, solubilization solution [10% sodium dodecyl sulfate (SDS) and 5% isopropanol in 12 mM HCl] was added, the cells were incubated at 37°C overnight, and the absorbance of the solution was measured at 570 nm.
Flow cytometry
For surface molecule staining, cells were incubated with anti-I-Ab-phycoerythrin (PE) (BD Biosciences Pharmingen, San Diego, CA), anti-CD40-fluorescein isothiocyanate (FITC) (Biolegend, San Diego, CA), anti-CD80-FITC (BD), anti-CD86-PE (Immunotech, Prague, Czech Republic), or anti-CD4-peridinin chlorophyll protein (PerCP) (BD) antibodies for 30 min at 4°C. Intracellular COX-1 and COX-2 were stained according to a previously described protocol (Ruitenberg and Waters, Citation2003). After the cells were washed with phosphate-buffered saline (PBS), they were fixed with Cytofix/Cytoperm (BD), washed, and incubated with anti-COX-1-PE and anti-COX-2-FITC antibodies (Cayman Chemical) for 30 min at 4°C. Isotype control antibodies were used to determine background fluorescence. The fluorescence intensity was measured using a flow cytometer (BD FACScan, BD Biosciences Immunocytometry Systems, San Jose, CA) equipped with a 488-nm argon laser; the filter settings for FITC, PE, and PerCP were 530 nm, 585 nm, and 650 nm, respectively. At least 10,000 cells per sample were collected for analysis.
Arachidonic acid (AA) release
DC (5 × 105 cells/well) were seeded into 24-well plates in serum-free RPMI containing 0.1 µCi [3H]-AA/ml and incubated for 4 h at 37°C. Cells were washed three times with 0.2% fatty acid-free bovine serum albumin (BSA-FAF; Sigma) in PBS. Cells were stimulated in RPMI/BSA-FAF (500 µl/well) with either LPS (1 µg/ml) or recombinant murine TNFα (10 ng/ml) for 5 h, and then 100 µl of each supernatant was transferred to 1 ml OPTI FLUOR (Perkin Elmer) and the radioactivity of the released AA was measured. After removal of the culture medium, the remaining cells were lysed with 500 µl of 2% Triton X-100, and the radioactivity in 100 µl of the lysate (in 1 ml OPTI FLUOR) was measured. The amount of AA released was expressed as a percentage and was calculated as follows: 100 × (released radioactivity)/(released radioactivity + radioactivity of lysed cells).
Western blot
DC were lysed in a lysis buffer (CelLytic™ M Cell Lysis Reagent; Sigma) containing a protease inhibitor cocktail (Roche Applied Science, Mannheim, Germany). In some experiments, splenic T-cells [isolated using magnetic beads (Pan-T-cell isolation kit, Milteny Biotec)] were lysed in the same manner. Cell lysates (10 µg/lane) were separated over a 10% SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) gel and transferred to a polyvinylidene fluoride membrane (Bio-Rad Laboratories, Hercules, CA). The membranes were incubated with rabbit anti-GABA-A receptor β2 antibody (1:1000; Thermo Scientific, IL) or rabbit anti-actin antibody (1:500; Abcam, Cambridge, UK). Proteins were visualized using horseradish peroxidase (HRP)-conjugated anti-rabbit antibody (1:10,000; Zymed Laboratories, South San Francisco, CA) and ECL-plus Western Blotting Detection Reagent (Amersham Biosciences, Buckingham, UK).
COX activity
For the total COX (COX-1 plus COX-2) activity assessment, DC were stimulated with LPS (1 µg/ml) for 18 h to induce COX-2 protein expression and then washed three times with PBS and cultured for 1 h with 10 µM AA in the presence of propofol, propofol analog (2,6-di-tert-butylphenol), or selective COX-2 inhibitor NS-398. The culture supernatant was harvested and PGE2 concentrations were measured. To assess COX-1 activity, LPS-non-stimulated DC were treated with 10 µM AA in the presence of NS-398 (5 µM) in the presence of propofol. After a 1-h culture, the supernatant was collected for PGE2 measurement.
Co-cultures and T-cell proliferation assay
DC were stimulated with LPS (1 µg/ml) for 48 h in the presence of either propofol or NS-398. Purified CD4+CD62L+ naïve T-cells (CD4+CD62L+ T-cell isolation kit II, Milteny Biotec) were obtained from the spleens of BALB/c mice and labeled with carboxyfluorescein succinimidyl ester (CFSE) (Dojindo, Kumamoto, Japan) according to the manufacturer’s protocols. Matured DC (C57BL6 origin) were seeded (104/well), together with CD4+CD62L+ T-cells (from BALB/c spleen) (105/well), into 96-well round-bottom plates. After 5 days of co-culture, T-cell proliferation was assessed, using flow cytometry, by determining the proportion of cells whose levels of CFSE decreased from that of T-cells in the T-cell-only culture (which served as negative control for proliferation). T-cells were identified with PerCP-conjugated anti-CD4-antibody-positive staining.
Statistical analysis
One-way analysis of variance (ANOVA) with Bonferroni post hoc test was conducted for statistical analysis using Graph Pad Prism 4 (GraphPad, San Diego, CA). Data were expressed as mean ± SEM. A p-value <0.05 was considered statistically significant.
Results
PGE2 and TXB2 production by DC
Upon LPS stimulation, PGE2 production increased significantly but was suppressed by propofol in a dose-dependent manner (). As expected, the selective COX-2 inhibitor, NS-398, inhibited PGE2 production to a similar degree as that observed without LPS. Propofol also suppressed the LPS-induced increase in TXB2 production. The inhibition was not due to cell toxicity because cell viability did not decrease in the presence of propofol (although cell viability decreased in the presence of LPS, irrespective of the presence of propofol), as indicated by the MTT assay.
Figure 1. (a) Prostaglandin (PG) E2 and thromboxane (TX) B2 production, and cell viability. Dendritic cells (DC) were stimulated with 1 µg lipopolysaccharide (LPS)/ml in the presence of propofol for 24 h, and PGE2 and TXB2 concentrations in the culture supernatants were then measured. NS-398 (a selective COX-2 inhibitor) was used at 5 µM (n = 6). Cell viability was assessed by MTT assay (n = 6). #p < 0.01, ##p < 0.001 vs no LPS. *p < 0.001 vs LPS/0 µM propofol. n.s. = not significant. (b) Cyclo-oxygenase (COX)-1 and COX-2 protein expression. DC were stimulated with 1 µg LPS/ml for 24 h in the presence of 30 µM propofol and then intracellular COX-1 and COX-2 protein expression was determined by flow cytometry. Representative dot-plots and summary figures are shown (n = 5). #p < 0.001 vs no LPS. n.s. = not significant. (c) Arachidonic acid (AA) release. DC were labeled with 0.1 µCi/ml [3H]-AA for 4 h and then stimulated with 1 µg LPS/ml in the presence of propofol for 5 h; thereafter, the amount of released AA was measured. As a positive control, a separate set of DC was stimulated with tumor necrosis factor (TNF)-α (10 ng/ml) for 5 h (n = 5). #p < 0.001 vs no LPS. n.s. = not significant.
![Figure 1. (a) Prostaglandin (PG) E2 and thromboxane (TX) B2 production, and cell viability. Dendritic cells (DC) were stimulated with 1 µg lipopolysaccharide (LPS)/ml in the presence of propofol for 24 h, and PGE2 and TXB2 concentrations in the culture supernatants were then measured. NS-398 (a selective COX-2 inhibitor) was used at 5 µM (n = 6). Cell viability was assessed by MTT assay (n = 6). #p < 0.01, ##p < 0.001 vs no LPS. *p < 0.001 vs LPS/0 µM propofol. n.s. = not significant. (b) Cyclo-oxygenase (COX)-1 and COX-2 protein expression. DC were stimulated with 1 µg LPS/ml for 24 h in the presence of 30 µM propofol and then intracellular COX-1 and COX-2 protein expression was determined by flow cytometry. Representative dot-plots and summary figures are shown (n = 5). #p < 0.001 vs no LPS. n.s. = not significant. (c) Arachidonic acid (AA) release. DC were labeled with 0.1 µCi/ml [3H]-AA for 4 h and then stimulated with 1 µg LPS/ml in the presence of propofol for 5 h; thereafter, the amount of released AA was measured. As a positive control, a separate set of DC was stimulated with tumor necrosis factor (TNF)-α (10 ng/ml) for 5 h (n = 5). #p < 0.001 vs no LPS. n.s. = not significant.](/cms/asset/1edb7cf1-2f9b-4f15-8534-6cd8c90c8d06/iimt_a_620036_f0001_b.gif)
Cyclo-oxygenase (COX) protein expression
Neither LPS nor propofol altered COX-1 expression (. LPS stimulation induced COX-2, whose expression was not altered by propofol. Propofol did not alter the percentages of COX-2+ cells after LPS stimulation.
AA release
The release of AA from DC was increased by LPS, but was unaffected by propofol (. As expected, TNFα stimulation also increased the release of AA.
COX activity
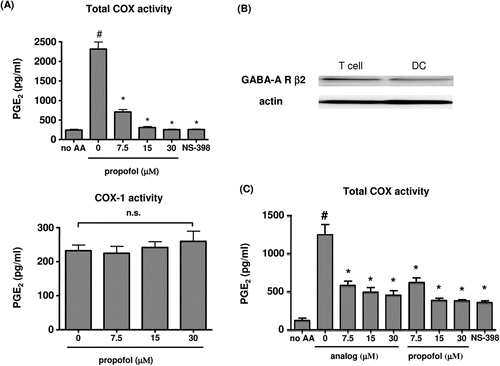
Total COX activity was significantly suppressed by propofol (), and the propofol-induced suppression at 30 µM was similar to the suppression observed with NS-398, a selective COX-2 inhibitor, leaving only COX-1 activity. In contrast, in non-stimulated cells in the presence of NS-398, propofol had no effect on PGE2 production, indicating that propofol does not affect COX-1 activity in the range of concentrations studied. In the DC, the GABA-A receptor β2 subunit was detected by Western blotting (. Propofol itself and the propofol analog significantly suppressed COX activity to a similar extent (.
Figure 2. (a) Cyclo-oxygenase (COX) activity. To determine total COX activity, DC were stimulated with 1 µg LPS/ml for 18 h, washed, and then incubated with 10 µM AA for 1 h. NS-398 was used at a dose of 5 µM (n = 6). To determine COX-1 activity, unstimulated DC were incubated with 10 µM AA in the presence of NS-398 for 1 h (n = 6). #p < 0.001 vs no AA, *p < 0.001 vs LPS/0 µM propofol. n.s. = not significant. (b) Western blotting to assess the presence of GABA-A β2 receptors on DC. As a positive control, GABA-A β2 receptors in total T-cells are depicted. Representative data from three independent experiments are shown. (c) Total COX activity after exposure to propofol analog 2,6-di-tert-butylphenol. DC were stimulated with 1 µg LPS/ml for 18 h, washed, and incubated with 10 µM AA for 1 h. NS-398 was used at a dose of 5 µM (n = 6). #p < 0.001 vs no AA. *p < 0.001 vs AA/0 µM propofol/analog.
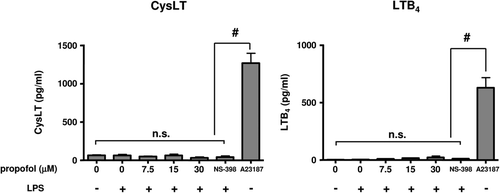
CysLT and LTB4 production by DC
The studies here showed that LPS treatment did not significantly up-regulate the production of either cysLT or LTB4. Propofol did not significantly affect either cysLT or LTB4 production by LPS-stimulated DC ().
Figure 3. Cysteinyl Leukotrienes (CysLT) and Leukotriene-B4 (LTB4) production. Dendritic cells (DC) were stimulated with 1 µg lipopolysaccharide (LPS)/ml in the presence of propofol for 24 h, and cysLT and LTB2 concentrations in the culture supernatants were then measured. NS-398 (a selective COX-2 inhibitor) was used at 5 µM. As a positive control for LT production, a separate set of DC was stimulated with a calcium ionophore, A23187 (5 μM) for 24 h (n = 5). #p < 0.001. n.s. = not significant.
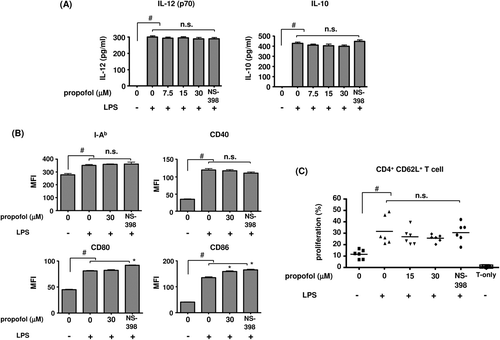
Production of IL-12 (p70) and IL-10, and expression of I-Ab and surface co-stimulatory molecules, after suppression of endogenous PGE2 production
LPS stimulation significantly increased the production of IL-12 (p70) and IL-10, which was not affected by either propofol or NS-398 (). The expression of all surface molecules tested was significantly up-regulated by LPS. I-Ab and CD40 expression was not altered by either propofol or NS-398. There were significant but small increases in CD80 and CD86 expression with NS-398, while CD86 expression was also slightly increased with propofol (.
Figure 4. (a) Interleukin (IL)-12 and IL-10 production. Measures were made of IL-12 and IL-10 formation by DC that were stimulated with 1 µg LPS/ml in the presence of propofol for 48 h. The selective COX-2 inhibitor NS-398 was used at 5 µM (n = 6). #p < 0.001 vs no LPS. n.s. = not significant. (b) DC surface markers after stimulation with 1 µg LPS/ml in the presence of propofol for 48 h (n = 6). #p < 0.001 vs no LPS. *p < 0.05 vs LPS/0 µM propofol. n.s. = not significant. (c) CD4+ CD62L+ naïve T-cell proliferation with DC co-culture. DC were stimulated with 1 µg LPS/ml in the presence of propofol or NS-398 (5 µM) for 48 h. The DC were then washed and co-cultured with CD4+CD62L+ naïve T-cells for 5 days. Percentages of proliferating T-cells were then determined by flow cytometry (n = 6). #p < 0.05 vs no LPS. n.s. = not significant.
Co-cultures and T-cell proliferation
With LPS stimulation, DC gained enhanced capability to push proliferation of CD4+CD62L+ T-cells (. LPS stimulation in the presence of propofol or NS-398 did not significantly affect the percentages of proliferating T-cells after the co-culture.
Discussion
The present study showed that propofol possesses a COX-inhibiting property in murine dendritic cells (DC). The mechanism of COX inhibition remains unknown, but might be related to the chemical structure of propofol, i.e., it is similar to that of γ-tocopherol and its major metabolite, 2,7,8-trimethyl-2-(β-carboxyethyl)-6-hydroxychroman (γ-CEHC) which are reported to inhibit COX-2 activity by a mechanism that also remains unknown (Jiang et al., Citation2000). In addition, this study found that endogenous suppression of COX activity in DC had little consequence in DC with respect to IL-12/IL-10 production, DC surface molecule expression, and allogeneic T-cell proliferation.
The presence of GABA-A receptors on T-cells (Alam et al., Citation2006; Bjurstom et al., Citation2008), monocytes (Alam et al., Citation2006), and microglias (Lee et al., Citation2011) has already been demonstrated, but its presence on DC has not been reported. We could find expression of the receptor protein in murine bone marrow-derived DC. We also examined whether propofol inhibits COX activity via the GABA-A receptor because propofol is known as a GABA-A receptor agonist in the central nervous system for exerting its anesthetic effect (Bali and Akabas, Citation2004). To this end, we used the propofol analog 2,6-di-tert-butylphenol which reportedly does not bind to the GABA-A receptor and is devoid of anesthetic effect (Ahrens et al., Citation2004). The analog also significantly suppressed COX activity to a similar extent as was observed with propofol. Therefore, the GABA-A receptor is unlikely to be related to the COX-inhibiting activity of propofol.
Exogenous PGE2 can act on DC to inhibit IL-12 production and stimulate IL-10 production (van der Pouw Kraan et al., Citation1995; Son et al., Citation2006; Sa-Nunes et al., Citation2007; Shiraishi et al., Citation2008). Therefore, we tested whether endogenous suppression of PGE2 production with propofol resulted in increased IL-12 production and decreased IL-10 production. However, in our study of murine DC, endogenous COX inhibition by propofol, as well as by NS-398, had no effect on either IL-12 or IL-10 production. Previous studies reinforced our findings in that endogenous COX inhibition had no effect on DC IL-12 or IL-10 production (Jozefowski et al., Citation2003; Yao et al., Citation2009). However, Harizi and colleagues (Harizi and Gualde, Citation2002; Harizi et al., Citation2002) have consistently reported that endogenous PGE2 inhibition in the presence of COX inhibitors increases IL-12 and decreases IL-10 production in DC. The reason for the conflicting results is unknown, but may be related to the level of surface PGE2 receptor expression and/or the maturation-activation status of DC (Tilley et al., Citation2001). Alternatively, the reason for the failure to alter cytokine production with COX inhibition may be that there was too little endogenous PGE2 production to suppress and enhance IL-12 and IL-10 production, respectively, prior to the addition of COX inhibitors.
Exogenous PGE2 has also been reported to inhibit expression of MHC Class II, CD40, and CD86 molecules (Gualde and Harizi, Citation2004; Sa-Nunes et al., Citation2007; Jung et al., Citation2010). Therefore, we wondered whether COX inhibition leads to up-regulation of these surface markers, resulting in an enhanced proliferation of helper T-cells by DC (Sa-Nunes et al., Citation2007). In the present study, both propofol and NS-398 were associated with a small but significant increase in CD86 expression (and NS-398 was also associated with an increase in CD80 expression). However, the up-regulation seems too small to enhance naïve helper T-cell proliferation. We assume that part of the reason for the small effect of COX inhibition on DC surface markers may come from the small amount of PGE2 produced by the DC that we generated. Indeed, far higher concentrations of PGE2 (e.g., µM range) have been employed to clearly demonstrate the effect of exogenous PGE2 on DC phenotype and function (Scandella et al., Citation2002; Baratelli et al., Citation2004).
When a drug is used to inhibit COX activity, AA cannot proceed through the COX pathway. Rather, it gets shunted into other enzymatic pathways, one of the most prominent of which may be the 5-lipoxygenase pathway, which leads to the production of leukotriens (LT). Thus, it is possible that propofol, via inhibition of COX, increases LT concentrations in the supernatants. The primary LT produced by DC are cysLT and LTB4 (Jozefowski et al., Citation2005). CysLT are potent mediators of asthma and hypersensitivity (Henderson, Citation1994; Byrum et al., Citation1999). They induce bronchoconstriction, increased microvascular permeability, and coronary artery vasoconstriction (Henderson, Citation1994; Byrum et al., Citation1999). On the other hand, LTB4 stimulates neutrophil functions, including aggregation, reactive oxygen species production, and chemotaxis (Henderson, Citation1994; Byrum et al., Citation1999). Furthermore, it is reported that LT can act on DC to modulate their migration and cytokine production (Robbiani et al., Citation2000; Machida et al., Citation2004; Jozefowski et al., Citation2005). In order to address these issues, cysLT and LTB4 concentrations in DC culture supernatants after LPS treatment were measured (). LPS treatment did not significantly up-regulate either cysLT or LTB4, which is consistent with a previous study (Jozefowski et al., Citation2005). Propofol significantly affected neither cysLT nor LTB4 production. Thus, the concern that propofol, via its COX-inhibiting property, could divert AA to produce an increased amount of LT does not appear to be an issue in our experimental setting.
Conclusions
The intravenous anesthetic propofol suppresses PGE2 production from dendritic cells (DC) by inhibiting COX enzyme activity. Endogenous COX inhibition with propofol did not significantly affect the production of IL-12 and IL-10, expression of I-Ab and co-stimulatory molecules (except for that of CD86, which was slightly increased), or the capacity to induce allogeneic T-cell proliferation. Therefore, it seems likely that the impact of COX-inhibition by propofol on DC function is minimal. However, it is not clear to what extent the results reported here would reflect activity in vivo—or are relevant for human health.
Declaration of interest
This work was supported by a Grant-in-AID for Scientific Research (C)-22591721 from the Japan Society for the Promotion of Science. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Ahrens J., Haeseler G., Leuwer M., Mohammadi B., Krampfl K., Dengler R., and Bufler J. 2004. butylphenol, a non-anesthetic propofol analog, modulates α1β glycine receptor function in a manner distinct from propofol. Anesth. Analg. 99:91–96.
- Alam S., Laughton D. L., Walding A., andWolstenholme A. J. 2006. Human peripheral blood mononuclear cells express GABAA receptor subunits. Mol. Immunol. 43:1432–1442.
- Bali M., andAkabas M. H. 2004. Defining the propofol binding site location on the GABAA receptor. Mol. Pharmacol. 65:68–76.
- Baratelli F. E., Heuze-Vourc’h N., Krysan K., Dohadwala M., Riedl K., Sharma S., andDubinett S. M. 2004. Prostaglandin E2-dependent enhancement of tissue inhibitors of metalloproteinases-1 production limits dendritic cell migration through extracellular matrix. J. Immunol. 173:5458–5466.
- Bjurstom H., Wang J., Ericsson I., Bengtsson M., Liu Y., Kumar-Mendu S., Issazadeh-Navikas S., and Birnir B. 2008. GABA, a natural immunomodulator of T-lymphocytes. J. Neuroimmunol. 205:44–50.
- Byrum R. S., Goulet J. L., Snouwaert J. N., Griffiths R. J., andKoller B. H. 1999. Determination of the contribution of cysteinyl leukotrienes and leukotriene B4 in acute inflammatory responses using 5-lipoxygenase- and leukotriene A4 hydrolase-deficient mice. J. Immunol. 163:6810–6819.
- Fogel-Petrovic M., Long J. A., Knight D. A., Thompson P. J., andUpham J. W. 2004. Activated human dendritic cells express inducible cyclo-oxygenase and synthesize prostaglandin E2 but not prostaglandin D2. Immunol. Cell Biol. 82:47–54.
- Gualde N., and Harizi H. 2004. Prostanoids and their receptors that modulate dendritic cell-mediated immunity. Immunol. Cell Biol. 82:353–360.
- Harizi H., and Gualde N. 2002. Dendritic cells produce eicosanoids, which modulate generation and functions of antigen-presenting cells. Prostagland. Leukotriene Essent. Fatty Acids 66:459–466.
- Harizi H., Juzan M., Pitard V., Moreau J. F., andGualde N. 2002. Cyclo-oxygenase-2-issued prostaglandin E2 enhances the production of endogenous IL-10, which down-regulates dendritic cell functions. J. Immunol. 168:2255–2263.
- Henderson W. R.Jr, . 1994. The role of leukotrienes in inflammation. Ann. Intern. Med. 121:684–697.
- Inada T., Kubo K., and Shingu K. 2010. Promotion of interferon-γ production by natural killer cells via suppression of murine peritoneal macrophage prostaglandin E production using intravenous anesthetic propofol. Int. Immunopharmacol. 10:1200–1208.
- Jiang Q., Elson-Schwab I., Courtemanche C., andAmes B. N. 2000. γ-Tocopherol and its major metabolite, in contrast to α-tocopherol, inhibit cyclo-oxygenase activity in macro- phages and epithelial cells. Proc. Natl. Acad. Sci. USA 97:11494–11499.
- Jozefowski S., Biedron R., Bobek M., and Marcinkiewicz J. 2005. Leukotrienes modulate cytokine release from dendritic cells. Immunology 116:418–428.
- Jozefowski S., Bobek M., and Marcinkiewicz J. 2003. Exogenous but not endogenous prostanoids regulate cytokine secretion from murine bone marrow dendritic cells: EP2, DP, and IP but not EP1, EP3, and FP prostanoid receptors are involved. Int. Immunopharmacol. 3:865–878.
- Jung I. D., Jeong Y. I., Lee C. M., Noh K. T., Jeong S. K., Chun S. H., Choi O. H., Park W. S., Han J., Shin Y. K., Kim H. W., Yun C. H., andPark Y. M. 2010. COX-2 and PGE2 signaling is essential for the regulation of IDO expression by curcumin in murine bone marrow-derived dendritic cells. Int. Immunopharmacol. 10:760–768.
- Lee M., Schwab C., andMcGeer P. L. 2011. Astrocytes are GABAergic cells that modulate microglial activity. Glia 59:152–165.
- Lutz M. B., Kukutsch N., Ogilvie A. L., Rossner S., Koch F., Romani N., andSchuler G. 1999. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Meth. 223:77–92.
- Machida I., Matsuse H., Kondo Y., Kawano T., Saeki S., Tomari S., Obase Y., Fukushima C., and Kohno S. 2004. Cysteinyl leukotrienes regulate dendritic cell functions in a murine model of asthma. J. Immunol. 172:1833–1838.
- Mak T., andSaunders M. E. (Eds.). 2006. The Immune Response: Basic and Clinical Principles. Burlington, VT: Elsevier Academic Press.
- Needleman P., Turk J., Jakschik B. A., Morrison A. R., andLefkowith J. B. 1986. Arachidonic acid metabolism. Annu. Rev. Biochem. 55:69–102.
- Norgauer J., Ibig Y., Gmeiner D., Herouy Y., andFiebich B. L. 2003. Prostaglandin E2 synthesis in human monocyte-derived dendritic cells. Int. J. Mol. Med. 12:83–86.
- Otto J. C., andSmith W. L. 1995. Prostaglandin endoperoxide synthases-1 and -2. J. Lipid Mediat. Cell Signal. 12:139–156.
- Rigouzzo A., Girault L., Louvet N., Servin F., De-Smet T., Piat V., Seeman R., Murat I., and Constant I. 2008. The relationship between bi-spectral index and propofol during target-controlled infusion anesthesia: A comparative study between children and young adults. Anesth. Analg. 106:1109–1116.
- Robbiani D. F., Finch R. A., Jager D., Muller W. A., Sartorelli A. C., andRandolph G. J. 2000. The leukotriene C4 transporter MRP1 regulates CCL19 (MIP-3β, ELC)-dependent mobilization of dendritic cells to lymph nodes. Cell 103:757–768.
- Ruitenberg J. J., andWaters C. A. 2003. A rapid flow cytometric method for the detection of intracellular cyclooxygenases in human whole blood monocytes and a COX-2-inducible human cell line. J. Immunol. Meth. 274:93–104.
- Sa-Nunes A., Bafica A., Lucas D. A., Conrads T. P., Veenstra T. D., Andersen J. F., Mather T. N., Ribeiro J. M., andFrancischetti I. M. 2007. Prostaglandin E2 is a major inhibitor of dendritic cell maturation and function in Ixodes scapularis saliva. J. Immunol. 179:1497–1505.
- Scandella E., Men Y., Gillessen S., Forster R., and Groettrup M. 2002. Prostaglandin E2 is a key factor for CCR7 surface expression and migration of monocyte-derived dendritic cells. Blood 100:1354–1361.
- Shiraishi H., Yoshida H., Saeki K., Miura Y., Watanabe S., Ishizaki T., Hashimoto M., Takaesu G., Kobayashi T., and Yoshimura A. 2008. Prostaglandin E2 is a major soluble factor produced by stromal cells for preventing inflammatory cytokine production from dendritic cells. Int. Immunol. 20:1219–1229.
- Sompayrac L. (Ed.). 2003. How the Immune System Works? Malden, MA: Blackwell Publishing.
- Son Y., Ito T., Ozaki Y., Tanijiri T., Yokoi T., Nakamura K., Takebayashi M., Amakawa R., and Fukuhara S. 2006. Prostaglandin E2 is a negative regulator on human plasmacytoid dendritic cells. Immunology 119:36–42.
- Tilley S. L., Coffman T. M., andKoller B. H. 2001. Mixed messages: Modulation of inflammation and immune responses by prostaglandins and thromboxanes. J. Clin. Invest. 108:15–23.
- van der Pouw Kraan T. C., Boeije L. C., Smeenk R. J., Wijdenes J., andAarden L. A. 1995. Prostaglandin-E2 is a potent inhibitor of human IL-12 production. J. Exp. Med. 181:775–779.
- Vanlersberghe C., and Camu F. 2008. Propofol. Handbook Exp. Pharmacol. 182:227–252.
- Yao C., Sakata D., Esaki Y., Li Y., Matsuoka T., Kuroiwa K., Sugimoto Y., and Narumiya S. 2009. Prostaglandin E2-EP4 signaling promotes immune inflammation through TH1 cell differentiation and TH17 cell expansion. Nat. Med. 15:633–640.