Abstract
As a new type of persistent organic pollutant, perfluorooctane sulfonate (PFOS) has raised great concern in recent years due to its ubiquitous distribution in the general environment and its long elimination half-life in humans. PFOS has toxic and carcinogenic effects in animals and humans, but the effects of PFOS on apoptosis are still not clear. The present study aimed to determine the mode of cell death and its mechanism in splenocytes and thymocytes from adult male C57BL/6 mice administered 0, 1, 5, or 10 mg PFOS/kg/day by gavage daily for 7 days. The results showed that more apoptotic cells were present in PFOS-treated mice than in control mice. PFOS induced production of reactive oxygen species (ROS), dissipation of mitochondria membrane potential, and apoptosis of splenocytes and thymocytes. Moreover, activities of superoxide dismutase, catalase, and glutathione reductase were increased, whereas activities of glutathione-S-transferase and glutathione peroxidase were decreased, in splenocytes. Glutathione contents were reduced as well. Differential expressions of proteins such as p53, Bax, caspase-3, and caspase-9 were significantly up-regulated in PFOS-exposed hosts, whereas Bcl-2 expression was significantly down-regulated. One possible mechanism for the findings here was that PFOS could overwhelm homeostasis of anti-oxidative systems, boost ROS generation, impact on mitochondria, and affect protein expression of apoptotic regulators, the latter of which resulted in initiation of the apoptosis program. Results from this study may provide a new insight into the potential adverse effects of PFOS exposure on humans, at the cellular level.
Introduction
As a new type of persistent organic pollutant (POPs), perfluorooctane sulfonate [PFOS; CF3(CF2)7SO3−] has aroused scientists’ great concern in recent years due to its ubiquitous distribution in the general environment (Giesy and Kannan, Citation2001; Lau et al., Citation2007; Sedlak and Greig, Citation2012) and its long elimination half-life in humans (mean is 8.7 years with a standard deviation of 6.1 years; range = 2.3–21.3 years) (Burris et al., Citation2002). Furthermore, it has been undergoing biological concentration and magnification, leading to excessive accumulation among the higher trophic levels of the food chain, such as predators and human beings (de Vos et al., Citation2008; Egeghy and Lorber, Citation2011; Noorlander et al., Citation2011). Food ingestion appears to be the primary route of exposure in the general population. Egeghy and Lorber (Citation2011) reported that the estimated average PFOS intake for adults ranged from 1.6–24.2 ng/kg of body weight per day. In our recent study conducted in the city of Shenyang, China, we found that the serum PFOS level in non-occupationally exposed individuals increased significantly from 0.03 µg/L in 1987 to 22.4 µg/L in 2002 (a 747-fold increase) and increased 13-fold just in the period from 1999–2002 (Jin et al., Citation2007).
PFOS has been added to Annex B of the CitationStockholm Convention on Persistent Organic Pollutants (2009) because it has had an increasingly negative effect on human health and on the environment. Animal studies have indicated that PFOS causes hepatotoxicity, developmental toxicity, reproductive toxicity, neurotoxicity, endocrine disruption, immunotoxicity and tumors of the liver and of the thyroid and mammary glands (Lau et al., Citation2007; OECD, 2002). It has been found that occupational exposure to PFOS is associated with an elevated cancer (such as bladder and prostate cancer) morbidity rate (Gilliand and Mandel, 1993; Alexander et al., Citation2003). Epidemiological investigations in the general population have revealed that elevated concentrations of serum PFOS are associated with thyroid disease, low birth weight, pre-eclampsia, impairment of semen quality, as well as an increased prevalence of attention-deficit hyperactivity disorder and infectious diseases in children (Joensen et al., Citation2009; Stein et al., Citation2009; Andersen et al., Citation2010; Fei et al., Citation2010; Hoffman et al., Citation2010; Melzer et al., Citation2010).
Reports concerning the immunotoxicity of PFOS in vitro and in vivo have appeared in the literature (Keil et al., Citation2008; Lefebvre et al., Citation2008; Peden-Adams et al., Citation2008; Dong et al., Citation2009; Qazi et al., Citation2009; Brieger et al., Citation2011; Mollenhauer et al., Citation2011; Wang et al., Citation2011; Zheng et al., Citation2011; Corsini et al., Citation2012; Dewitt et al., Citation2012). However, there is still a lack of understanding of the toxicokinetics and mode(s)/mechanism(s) of immunotoxic action of PFOS. During the last decade, at the cellular level, PFOS has been shown to induce different biochemical changes, several of which are typically associated with apoptosis (Liu et al., Citation2007; Chen et al., Citation2011; Kim et al., Citation2011). Therefore, PFOS toxicity is thought to be caused, in part, by the induction of apoptosis.
Apoptosis plays an important role in development and homeostasis in vertebrates, and it is also a vital regulatory process of the immune system. Many lymphocytes undergo apoptosis at the termination of the acute phase of an immune response. This active regulatory mechanism could be a potential target for immunotoxicants that are known to destroy splenic and thymic tissues. In the present study, we investigated whether oral exposure to PFOS triggered the apoptosis of thymocytes and splenocytes and if it altered the expression of apoptosis-regulating molecules such as bcl-2, bax, p53, caspase-3, and caspase-9 at the protein level.
Methods
Chemicals, antibodies, and supplies
Perfluorooctane sulfonic acid potassium salt (PFOS; purity > 98%) was purchased from Fluka (Steinheim, Switzerland). Potassium PFOS suspensions were prepared in de-ionized water with 2% Tween®-80 at various concentrations. RPMI 1640 medium (containing L-glutamine and sodium bicarbonate), phosphate-buffered saline (PBS, pH 7.4; with or without Ca2+ and Mg2+), and penicillin/streptomycin were purchased from Cellgro (Mediatech, Herndon, VA). Fetal bovine serum (FBS) was purchased from Hyclone (Logan, UT). Dimethyl sulfoxide (DMSO, 99.9% purity) was purchased from Amresco (Solon, OH). Luma Plate, Unifilters, and Microscint 20™ were obtained from Packard (Meriden, CT); Triton-X, tissue culture plates, and disposable labware were purchased from Sigma (St Louis, MO). Sodium carbonate (> 99.5% pure) was obtained from Kanto Chemical (Tokyo, Japan). Methyl tertiary-butyl ether (MTBE; HPLC grade), methanol (HPLC grade), acetonitrile (HPLC grade), and ammonium acetate (> 97%) were purchased from Wako Pure Chemical Industries (Osaka, Japan).
Propidium iodide (PI) and an Annexin V-FITC Apoptosis Detection kit were purchased from BD Pharmingen (Frankin Lakes, NJ). RNase A, Rhodamine (Rh)-123, and 2,7-dichloro-fluorescein-diacetate (DCFH-DA) were purchased from Sigma. Anti-oxidative enzyme detection kits (SOD, CAT, GPx, GR, GST) and a kit to measure GSH were purchased from Nanjing Jian-cheng Bioengineering Institute (Nanjing, China). Polyclonal anti-Bcl-2, -p53, -Bax, -Caspase-3, and -Caspase-9 antibodies, along with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse antibody, were bought from Santa-Cruz Biotechnology (Santa Cruz, CA).
Animals
All experiments performed were reviewed and approved by the China Medical University Institutional Animal Ethics Committee and conformed to NIH guidelines on the ethical use of animals. For the experiments, adult male C57Bl/6 mice (8–10-weeks-old; weighing 18–20 g) were purchased from the Shanghai Laboratory Animal Center of Chinese Academy of Science (SLAC, Shanghai, China). Male mice were chosen to avoid the hormonal changes associated with cyclic ovulation/menstruation that can influence the immune system. Moreover, Peden-Adams et al. (Citation2008) demonstrated that male mice are more sensitive to PFOS immunotoxicity than same-strain females. Forty-eight mice were then randomly divided by body weight (BW) into four groups of 12 mice/group. Once distributed, the mice were acclimated to cage conditions and the treatment room (12-h light/dark cycle, 22 ± 2°C, 60–65% relative humidity) for 1 week before dosing was initiated. Throughout the studies, the mice were provided access to filtered water and food ad libitum.
Treatment regimens
Exposures consisted of oral administration of PFOS delivered in de-ionized water containing 2% Tween®-80. Control mice received deionized water containing 2% Tween®-80 only. Mice were dosed once daily via oral gavage for 7 days (0, 1, 5, or 10 mg/kg BW/day). Individual food intake (12 mice in each group were assigned to four cages [three/cage]) was calculated from total group consumption over each 24-h period divided by three. The body weight of each mouse was measured daily over the 7-day treatment period. Gavage volume was adjusted based on most current body weight (range between 100–130 µl) to assure consistency of the dose being delivered. Bedding, food, and water were changed twice a week and the mice were observed daily. The highest dose of PFOS used in this study (i.e., 10 mg/kg/day) resulted in a serum level of 126.49 mg/L, which was ~ 2–10-fold higher than what has been reported as the upper range of occupational exposures (Organization for Economic Cooperation and Development) (OECD, 2002).
Serum sampling, organ mass, and spleen cell culture
Mice were euthanized 24 h after the final exposure (i.e., Day 8 of study) via CO2 asphyxiation, and the blood was collected by cardiac puncture. The blood was centrifuged (400 × g, 4°C) for 10 min, and the sera were collected and placed at −80°C for later PFOS analysis. The spleen, thymus, liver, and kidneys were then collected aseptically and weighed. Organ mass was normalized by body weight and reported as the organ index ([organ weight/body weight] × 100).
The isolated spleen from each mouse was passed through a sterile fine-wire mesh with 10 ml of complete RPMI 1640 (media bearing 5% heat-inactivated FBS, 25 mM HEPES, 0.12% gentamicin, and 2 mM glutamine). Cell suspensions were centrifuged at 350 × g (10 min, room temperature [RT]); erythrocytes in the suspension were then lysed with cold 0.17 M ammonium chloride (NH4Cl), and the remaining cells were washed twice with the medium. Spleen cells were finally re-suspended in RPMI 1640 and then counted and checked for viability using a trypan blue exclusion assay (note: always > 90%). The cells were then diluted further with RPMI or (depending on experiment) aliquots removed, centrifuged, and re-suspended in PBS at the designated concentrations as outlined in the protocols below.
Serum PFOS analysis
Details of the analytical procedure to measure PFOS in each serum sample have been provided previously (Hansen et al., Citation2001; Zheng et al., Citation2011). Briefly, 0.5 ml of serum, 1 ml of 0.5 M tetrabutyl-ammonium hydrogen sulfate solution, and 2 ml of sodium carbonate buffer (0.25 M, pH 10) were added to 15 ml polypropylene tube and thoroughly mixed. Following addition of 5 ml MTBE, the organic and aqueous layers were separated by centrifugation, and the organic layer was removed. The aqueous mixture was rinsed with MTBE and separated twice. The solvent was evaporated at RT under an N2 gas flow, and the sample reconstituted in 0.5 ml methanol. The sample was then passed through a nylon filter (Autovial R5 PUNYL; 0.45-µm pore size; Whatman, Tokyo) to remove any suspended materials and insoluble particles. Each extracted solution was then analyzed by liquid chromatography-mass spectrometry (LC/MS system, Shimadzu, Kyoto, Japan) as previously described (Jin et al., Citation2007; Zheng et al., Citation2011).
Serum corticosterone analysis
On blood collection days, movement about the animal suite was limited. Animals were euthanized by dose group (to minimize any disturbance to other groups) and blood was collected via cardiac puncture within 5 min of removal from the cage. Blood was processed as described above for later analysis of corticosterone. Corticosterone levels in thawed serum samples were measured (in duplicate) using an RIA kit (ICN Biomedical, Costa Mesa, CA) with a 0.2 ng/ml sensitivity as described previously (Francis et al., Citation2000).
Apoptotic DNA fragmentation and cell death analysis
The DNA fragmentation (histone associated mono- and oligonucleosomes) was measured using a Cell Death Detection Sandwich ELISA kit (Roche, Germany). Samples of thymocytes and splenocytes (105 cells/ml) were each centrifuged (200 × g, 10 min), and the respective cell pellets were suspended in kit incubation buffer for 30 min at 15–25°C for lysis. Following centrifugation (20,000 × g. 10 min), each supernatant was diluted 10-fold with incubation buffer, and the nucleosomes in each sample solution then measured by ELISA. Modular microtitre plates were incubated overnight with 100 µl anti-histone antibody (coating solution) at 4°C. After aspirating the coating solution, 200 µl incubation buffer was added and allowed to stand for 30 min at 15–25°C. The plates were then rinsed with washing buffer (200 µl) three times, 100 µl of sample solution was added into each well, and the plate incubated for 90 min at 15–25°C. After washing, 100 µl conjugate solution (anti-DNA-peroxidase) was added to each well and the cells were incubated a further 90 min at 15–25°C. After the wells were washed, substrate ABTS (2-2′-azino-di- [3-ethylbenzthiazoline sulfonate]; 100 µl) was then added and incubated for 10 min before the absorbance was measured at 405 nm using an ELISA plate reader (ELx808 BIO-TEK Instruments, Winooski, VT).
Cell cycle analysis
Cells with hypo-diploid DNA were determined by cell cycle studies. The thymocytes and splenocytes (106 cells/ml) were each suspended in PBS, fixed by drop-wise addition of ice-cold 70% ethanol, and stored at 4°C overnight. The fixed cells were harvested, washed with PBS, and suspended in 1 ml PBS. Phosphate citrate buffer (200 µl, pH 7.8) was then added and the cells incubated for 60 min at RT. After centrifugation, the cells were re-suspended in 0.5 ml of PI stain (10 mg PI, 0.1 ml Triton-X 100, and 3.7 mg EDTA in 100 ml PBS) and 0.5 ml of RNase A (50 µg/ml) solution, and further incubated for 30 min in the dark. The PI fluorescence was then measured through a FL-2 filter (585 nm) and 10,000 events were acquired (Darzynkiewicz et al., Citation1992).
Assessment of apoptotic and necrotic cells
Thymocytes and splenocytes isolated from mice treated with PFOS for 7 days were each adjusted to 106 cells/ml in RPMI 1640 containing 10% heat-inactivated FBS. Apoptotic and necrotic cell distribution was analyzed by Annexin V binding and PI uptake. Positioning of quadrants on Annexin V/PI dot plots was performed, and living cells (Annexin V−/PI−), early apoptotic/primary apoptotic cells (Annexin V+/PI−), late apoptotic/secondary apoptotic cells (Annexin V+/PI+), and necrotic cells (Annexin V−/PI+) were distinguished (Vermes et al., Citation1995). Therefore, the total apoptotic proportion included the percentage of cells with fluorescence Annexin V+/PI− and Annexin V+/PI+.
The cells were suspended in 1 ml binding buffer (1×) and a 100 µl aliquot was incubated with 5 µl Annexin V-FITC and 10 µl PI for 15 min in the dark at RT, and 400 µl binding buffer (1×) was added to each sample. The FITC and PI fluorescence were measured through FL-1 (530 nm) and FL-2 (585 nm) filters, respectively, in a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA). A minimum of 10,000 events/sample were acquired. All data acquisition and analyses were performed utilizing CELLQuest software (Becton Dickinson).
Flow cytometric analysis of the expression of apoptosis-associated molecules
PFOS-induced changes in the percentage of cells expressing various apoptosis-associated molecules were evaluated by FACS analysis. For the determination of the expression of Bcl-2, p53, Bax, Caspase-3, and Caspase-9 proteins, splenic lymphocytes (106 cells/ml) were permeabilized and then fixed in 2% paraformaldehyde for 15 min followed by 70% ice-cold methanol for 1 h. Splenic lymphocytes were incubated with antibodies against Bcl-2, p53, Bax, Caspase-3, and Caspase-9 (0.05–0.8 µg/ml) for 30 min at RT. Cells expressing the various molecules were detected by incubation with FITC-conjugated secondary antibodies (50 ng/ml) for 30 min in the dark. Cells were washed thoroughly, and then 10,000 cells/sample were analyzed using FACS. The level of fluorescence intensity was used to estimate the percentages of cells expressing each of the various molecules.
Mitochondrial membrane potential (MMP)
For the detection of mitochondrial Δψ, the thymocytes and splenocytes (at 106/ml) were each incubated with 5 µg Rh-123/ml (60 min, in the dark, 37°C), then harvested and suspended in PBS. The mitochondrial Δψ was measured by the fluorescence intensity (FL-1, 530 nm) of 10,000 cells using the Becton Dickinson flow cytometer (Bai et al., Citation1999).
Reactive oxygen species (ROS) measurement
Generation of ROS was detected by DCF fluorescence. The thymocytes and splenocytes (at 106/ml) were each incubated with 100 µM DCFH-DA for 60 min in the dark at 37°C. The cells were then harvested, suspended in PBS, and ROS generation was measured by the fluorescence intensity (FL-1, 530 nm) of 10,000 cells using the Becton Dickinson flow cytometer (Wang et al., Citation1996).
Activities of anti-oxidant enzymes and glutathione (GSH) content
Splenocytes from mice treated with PFOS for 7 days were suspended in 1 ml PBS (106 cells/ml), then lysed by freezing at −80°C and rapid thawing at 4°C; this final step was repeated four times. The homogenate was then centrifuged (10,000 × g, 15 min, 4°C) to separate insoluble materials. The supernatant was removed and total protein content was assessed using the GSH assay kit. Thereafter, the material was assayed for superoxide dismutase (SOD), catalase (CAT), glutathione-reductase (GR), -peroxidase (GPx), and -S-transferase (GST) activities and GSH content using commercial kits, according to manufacturer protocols. SOD, CAT, GST, and GPx activities were ultimately expressed as U/mg protein, that of GR as U/g protein, and GSH content as mg GSH/g protein.
Statistics
All experiments were repeated twice (two trials), with n = 6 each. Experimental trials were tested for trial by treatment interactions and, when statistically possible, data from trials were combined. Data were tested for normality (using Shapiro-Wilks W-test) and homogeneity (using Bartlett’s test for unequal variances) and, if needed, appropriate transformations were made. The transformations (when required) are outlined in the figure legends. A one-way analysis of variance (ANOVA) was used to determine differences among doses for each end-point using SAS software (Version 9.13; SAS Institute Inc., Cary, NC) in which the standard error used a pooled estimate of error variance. When significant differences were detected by the F-test (p < 0.05), the Dunnett’s t-test was used to compare treatment groups to the control group.
Results
Animal body weight, food intake, organ indices, and serum PFOS
No significant differences in body weight, splenic, thymic, and kidney indices were found among the treatment groups over the 7-day study period (). On the last day of the experiment, food consumption was reduced in comparison to untreated mice only in the group exposed to 10 mg PFOS/kg/day. Furthermore, the hepatic indices of mice exposed to 5 or 10 mg PFOS/kg/day were increased by 23% and 52% above the levels for the control values, respectively. There was a dose-related increase in serum PFOS levels in the PFOS-exposed mice, but the corticosterone levels in these mice were not markedly affected ().
Table 1. Effects on serum corticosterone, body weight, and organ indices in mice treated orally with PFOS daily for 7 days.
Effect of PFOS on apoptotic and necrotic cells
Apoptotic thymocytes and splenocytes from mice treated with PFOS were examined by flow cytometry. The number of apoptotic cells increased in a dose-related manner (). A significant increase in hypo-diploid DNA, i.e., the sub G1 population, was observed in the spleen at ≥ 5 mg PFOS/kg/day dose and in the thymus at a dose of 10 mg PFOS/kg/day. The Annexin V binding assay measured fluorescence generated by Annexin binding externalized phosphatidyl-serine of apoptotic cells (which also incorporates PI) and also reflected upon necrotic cells. In the spleen and thymus, the number of total (early + late) apoptotic cells was enhanced progressively with dose and a substantial rise was noted at ≥ 5 mg PFOS/kg/day (). The distribution of the necrotic cells showed a substantial increase in cell necrosis induced by PFOS in thymocytes only. Surprisingly, there were no significant changes in the incidence of necrosis in splenocytes across all treatment groups.
Table 2. Distribution apoptotic and necrotic cells in spleens from mice treated daily with PFOS orally for 7 days.
Effect of PFOS on DNA damage
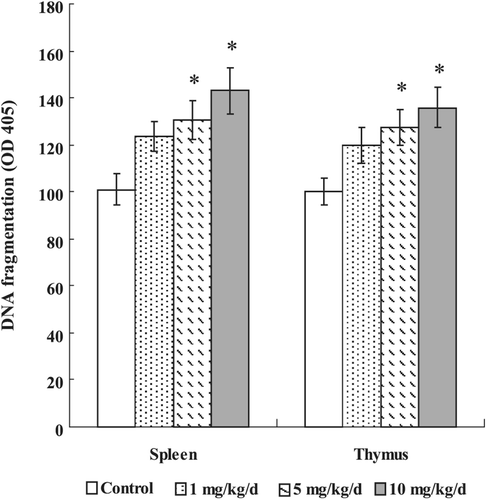
The sandwich ELISA method for histone-associated DNA fragments in the cell lysates of splenocytes and thymocytes from PFOS-treated mice revealed enhanced absorbance in a dose-dependent manner (). Compared with the control group (100.79 [± 6.67] in splenocytes, and 100.08 [± 5.53] in thymocytes), significantly higher DNA fragmentation (based on OD405 nm values) was observed at a dose of 5 mg PFOS/kg/day (130.45 [± 8.31] in splenocytes, 127.46 [± 7.36] in thymocytes) and 10 mg PFOS/kg/day (142.93 [± 9.84] in splenocytes, 136.12 [± 8.78] in thymocytes). The DNA fragments data of spleen and thymus matched well with the apoptotic data and confirmed apoptosis induced by in vivo PFOS.
Figure 1. Effects of PFOS on DNA fragmentation (mono- and oligonuclesomes). Thymocytes and splenocytes were isolated from male C57Bl/6 mice following oral exposures to PFOS for 7 days. DNA fragmentation was determined by Cell Death Detection ELISA. Data are presented as mean (± SE); n = 12/group. When significant differences were detected by the F-test (p < 0.05), a Dunnett’s t-test was used to compare treatment groups to the control group. *Value significantly different from control at p ≤0.05.
Effect of PFOS on MMP
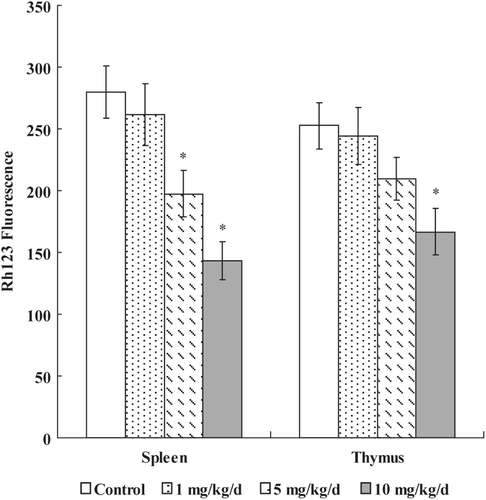
To determine if cell apoptosis induced by PFOS involved mitochondrial pathways, we measured the effects of PFOS exposure on MMP. As shown in , mitochondrial Δψ reduced substantially in splenocytes at doses of ≥ 5 mg PFOS/kg/day. In the case of thymocytes, mitochondrial depolarization was markedly observed only at dose of 10 mg PFOS/kg/day (166.42 [± 18.70]) as compared to that in the control group (252.63 [± 17.56] mean Rh-123 fluorescence units).
Figure 2. Effects of PFOS on mitochondrial membrane potential. Thymocytes and splenocytes were isolated from male C57Bl/6 mice following oral exposures to PFOS for 7 days. Rh-123 was added and the cells were incubated for 60 min. Fluorescence was measured using a flow cytometer with an FL-1 filter. Data are presented as mean (± SE); n = 12/group. When significant differences were detected by the F-test (p < 0.05), a Dunnett’s t-test was used to compare treatment groups to the control group. *Value significantly different from control at p ≤ 0.05.
Effect of PFOS on ROS
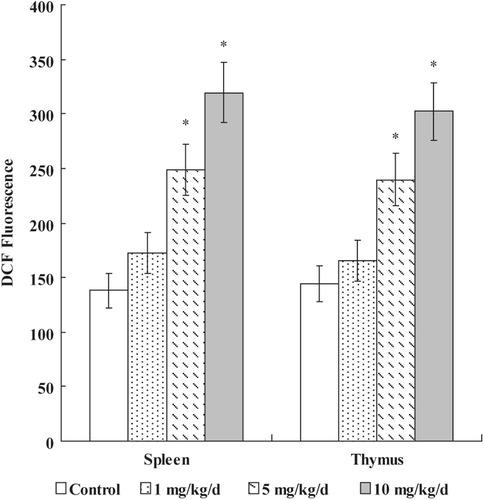
The DCF fluorescence proportionate to the ROS levels in the cells was monitored by flow cytometry. As shown in , after 7 days of exposure to PFOS, a significant increase of ROS was exhibited at ≥ 5 mg PFOS/kg/day dose in both spleen (5 mg PFOS/kg/day = 248.61 [± 23.11], 10 mg PFOS/kg/day = 319.37 [± 25.97], Control = 137.86 [± 15.41] mean DCF fluorescence units) and thymus (5 mg PFOS/kg/day = 239.76 [± 24.06], 10 mg PFOS/kg/day = 302.13 [± 25.97], Control = 144.08 [± 16.33]).
Figure 3. Effects of PFOS on generation of ROS. Thymocytes and splenocytes were isolated from male C57Bl/6 mice following oral exposures to PFOS for 7 days. DCFH-DA was added and the cells were incubated for 60 min. DCF fluorescence was measured using a flow cytometer with an FL-1 filter. Data are presented as mean (± SE); n = 12/group. When significant differences were detected by the F-test (p < 0.05), a Dunnett’s t-test was used to compare treatment groups to the control group. *Value significantly different from control at p ≤ 0.05.
Effect of PFOS on anti-oxidative enzyme activities and GSH content
After 7 days of exposure to PFOS, activities of anti-oxidative enzymes and the GSH content in splenocytes isolated from adult male C57Bl/6 mice were measured. As shown in , the GSH level and GPx activity were significantly reduced at ≥ 5 mg PFOS /kg/day dose, whereas SOD and GR activity were significantly increased in the 5 and 10 mg PFOS /kg/day exposure groups. CAT activity was only markedly increased at the high concentrations (10 mg/kg/day) in the PFOS treatment group, and the GST activity only decreased at a dose of 10 mg PFOS /kg/day.
Table 3. Anti-oxidative enzyme activity and GSH content in splenocytes from mice treated daily with PFOS orally for 7 days.
Effects of PFOS on p53, Bax, Bcl-2, caspase-3, and caspase-9 expression
showed the effects of PFOS on expression of bcl-2, p53, bax, caspase-3, and caspase-9 proteins in splenic lymphocytes, respectively. The results indicated that Bcl-2 protein expression was down-regulated, compared to the control, after PFOS exposures. In contrast, the expressions of p53, Bax, Caspase-3, and Caspase-9 were each enhanced—significantly so at the higher doses used—in a manner somewhat reflective of a dose-trend.
Table 4. Expression of bcl-2, p53, bax, caspase-3, and caspase-9 proteins in splenic lymphocytes from mice treated daily with PFOS orally for 7 days.
Discussion
Experimental evidence has indicated that the immune system is a major target for toxic effects of xenobiotics (Luster and Rosenthal, Citation1993). As such, adult male C57Bl/6 mice exposed to PFOS via oral gavage could serve as a model for studying the toxicity and toxic mechanisms of this emerging chemical of concern. Our results clearly showed that exposure to PFOS could potentiate reactive oxygen species (ROS) production and corresponding anti-oxidative responses in splenic and thymic cells from mice treated with PFOS, resulting in oxidative damage, as exemplified by DNA damage. PFOS exposure also enhanced p53, Bax, and caspase(s) expression, seemingly indicating that these are involved in the observed PFOS-induced apoptosis.
In this study, we evaluated body weight gain, food intake, weights of lymphoid organs, and serum corticosterone concentration of adult C57Bl/6 mice exposed to PFOS. Compared with untreated mice, the relative weights of splenic and thymic tissues were not significantly decreased in mice exposed to PFOS. In contrast, Wang et al. (Citation2011) conducted a study to explore the underlying mechanism of the immune organ atropy in mice that were fed a regular or high-fat diet and then exposed to PFOS (0, 5, and 20 mg/kg/day) for 14 days. Their results showed that body weight significantly decreased and the immune organs showed considerable atrophy in either the regular or high-fat diet group. However, consistent with our results, Lefebvre et al. (Citation2008) measured changes in immune tissues in adult rats exposed to PFOS for 28 days at levels ranging from 2–100 mg PFOS/kg diet (corresponding to ≈ 0.14–7.58 mg/kg/day); their results showed that the relative spleen and thymus weights were not significantly altered in male rats. As such, based on the above results, we believe the relative spleen and thymus weights might be affected not only by the PFOS exposure levels but also exposure times and animal species. Furthermore, our results revealed significant effects on ROS formation and apoptosis in the absence of significant splenic and thymic changes; this particular outcome may be highly significant from a mechanistic point of view.
Results of the present study also showed that exposure to 10 mg PFOS/kg/day via gavage for 7 days statistically reduced food consumption and increased liver mass. These results were consistent with similar observations in previous studies evaluating PFOS, perfluoroctanic acid (PFOA), and perfluorinated insecticide N-ethy1-perfluorooctane sulfonamide (N-EtPFOSA) (Peden-Adams et al., Citation2008; Qazi et al., Citation2009; Kim et al., Citation2011; Zheng et al., Citation2011; Elcombe et al., Citation2012). For example, a recent study by Elcombe et al. (Citation2012) evaluated PFOS-induced liver-related effects in male Sprague Dawley rats; following a 7-day dietary exposure to K+PFOS at 20 or 100 ppm, there was a significantly increased liver weight. Kim et al. (Citation2011) also obtained similar results at comparable doses in Sprague-Dawley rats. Those studies also indicated that the increased liver weight might be partially associated with reductions in body weight, since body weight and food intake significantly decreased at higher doses of PFOS. Our data showed that food consumption and body weight of the mice exposed to 10 mg PFOS/kg/day were 29% and 57%, respectively, less than those of the untreated mice. As such, the hepatic index increases may be partially due to decreased body weight, even though changes in that parameter were not statistically significant.
Inhibition of food intake is known to cause nutrient deficiency and malnutrition that are associated with impaired immune responses. The aspects of immunity most consistently affected by malnutrition are cell-mediated immunity, phagocyte function, the complement system, secreted antibodies, and protein production (Weller, Citation2001). In addition, moderate caloric restriction is known to reduce thymic and splenic weight and cellularity (Hosea et al., Citation2004), and alter the nucleolar structure and function in lymphocytes (Berger et al., Citation2005). Qazi et al. (Citation2009) recently reported that dietary treatment of mice with 0.02% PFOS and PFOA for 10 days (at serum PFOS levels 340 ± 16 mg/L) can reduce the total number of circulating white blood cells (leukopenia) and lymphocytes (lymphopenia), whereas the number of circulating neutrophils, the key cellular mediators of the innate immune response, was decreased only by PFOA. That study also suggested that PFOA induces neutropenia indirectly by the induction of severe food restriction. These effects of PFOS on the immune function are likely to be partially mediated via food restriction. However, our study also showed that, compared to the control group, neither food intake and body weight nor the serum corticosterone concentration were significantly different, but the anti-oxidative enzymes activity, production of ROS, and expression of p53 proteins in splenic lymphocytes were significantly altered in the group treated with 5 mg PFOS/kg/day, a dose that results in a serum level of 87.56 mg/L. This concentration is ≈ 20–100-fold greater than the concentration of PFOS reported in the sera of occupationally-exposed humans (OECD, 2002).
In immune cells, the oxidant–anti-oxidant balance is an important determinant of function and ROS act as potent signaling molecules in various cellular processes. ROS can be generated at many different organelles including mitochondrion, which is responsible for aerobic respiration, the most common source of ROS generation (Kelly et al. Citation1998). Pulido and Parrish (Citation2003), in their review paper, stated that excessive ROS generation can ultimately lead to apoptosis. Because mitochondria are the main target for damage by ROS, leading to necrosis and apoptosis as shown by Netto et al (2002), it is reasonable to assume that mitochondria may play a crucial role in PFOS-induced toxicity. The excessive ROS may cause mitochondria further damage and result in loss of the membrane potential, thus inducing apoptosis. The mitochondrial toxicity of perfluorinated chemicals (PFC) has been suggested as one of their mechanisms of toxicity, suggested as a subcellular target potentially mediating cytotoxicity. For example, the structure of PFOA is similar to a long-chain fatty acid, and therefore is capable of inducing change of the mitochondrial permeability transition (MPT) (Panaretakis et al., Citation2001). Also, O’Brien and Wallace (Citation2004) reported that N-EtPFOSA, the terminal metabolite of which is PFOS in vivo, could induce alternation of MPT and cause the release of cytochrome c, leading to inhibition of respiration and ROS generation. Therefore, based on our results in combination with those described above, it may be that PFOS exerts its toxic effects through ROS-mediated apoptosis, and that this involves the mitochondria.
In the present study, SOD activity was significantly increased with increased ROS production, and presumably to counteract oxidative damage at higher concentrations of PFOS (5 and 10 mg/kg/day). A significant CAT induction was observed at 10 mg PFOS/kg/day. CAT is mainly located in peroxisome and is responsible for the reduction of hydrogen peroxide and protection from the oxidation of unsaturated fatty acids in cell membrane (Livingstone, Citation2001). Therefore, the higher activity of CAT can be attributed to the increase of pro-oxidants. GST was significantly reduced and strongly inhibited by the high PFOS concentration (10 mg/kg/day). A decrease in GST may suggest a failure in detoxification and occurrence of oxidative stress in the cells (Santos et al., Citation2004). In this study, the decrease in GST activity was concomitant to the decrease in GSH content. Similarly, increase of GSH content can be viewed as an adaptation to low level of oxidative stress or failure of adaptation due to a severe oxidative stress, leading to suppressed GSH content and detoxification of oxidized GSH (GSSG) to GSH. ROS could be reduced by oxidizing GSH to GSSG, which is catalyzed by GPx. GPx catalyzes the reduction of hydrogen peroxides and is considered as an efficient protective enzyme against lipid peroxidation (Meyer et al., Citation2003). Indeed, a dose-dependent reduction of GPx activity was observed with PFOS exposure, suggesting an adaptive response to maintain the homeostasis of reduced GSH. GR activity significantly increased in the present study. GR is supposed to maintain cytosolic concentration of GSH by reducing GSSG to GSH. Hence, an increase in GR activity indicated compensation of cellular GSH depletion. In total, changes in activities of anti-oxidative enzymes following PFOS exposure indicated that PFOS may overwhelm the homeostasis of anti-oxidative systems, boost the generation of ROS, impact mitochondria, and result in initiation of an apoptosis program.
PFOS exposure caused significant up-regulation of p53 protein expression and significant down-regulation of Bcl-2 protein expression in splenic lymphocytes of adult male C57Bl/6 mice treated daily with PFOS. DNA damages in the thymocytes and splenocytes were also revealed by flow cytometric analysis. The tumor suppressor gene p53 has been examined for its role in DNA damage-induced apoptosis that is chemically induced (Langheinrich et al., Citation2002). Typically, p53 is activated when DNA damage occurs or the cell is stressed. It translocates to the nucleus, where it can induce pro-apoptotic gene expression, such as Bax, on the mitochondrial membrane, and to activate the effector caspase-3 and accelerate cell death (Sheikh and Fornace, Citation2000). Our results showed that the expression level of Bax progressively increased when the mice were treated with PFOS. Two previous studies have reported an increase in caspase-3 and caspase-9 activity and apoptosis in cultured tilapia hepatocytes after PFOS exposure (Liu et al., Citation2007; Hu XZ and Hu DC, 2009). Taking these results together, it may be hypothesized that PFOS-induced apoptosis in splenocytes might occur via an activated p53 that triggers the trans-activation of Bax and activates caspases-3 and -9, which then cleaves the death substrates, leading to apoptosis in splenocytes of mice treated with PFOS.
It is worth noting that the dose used in the present study resulted in serum levels of PFOS ranging from 24–126 mg/L. These levels are approximately (at least) 1000-times greater than the reported mean blood levels in the general population (i.e., 22.4 μg/L) (Jin et al., Citation2007). Some studies have indicated that the effects of PFOS on select immunological indices were not dose-related (Peden-Adams et al., Citation2007, Citation2008; Keil et al., Citation2008; Dong et al., Citation2009). For example, Keil et al. (Citation2008) studied effects of gestational exposure to PFOS on immune functions in mice and found that natural killer (NK) cell activity decreased 43% in male offspring after a 1 mg PFOS/kg/day treatment for 17 days; however, in offspring mice that had been exposed to 5 mg PFOS/kg/day, NK cell activity decreased only 32%. In our previous studies (Dong et al., Citation2009), adult male C57BL/6 mice were exposed to PFOS daily via gavage for 60 days (at 0, 0.5, 5, 25, 50, or 125 mg/kg total administered dose (TAD)); it was again found that there was a non-linear relationship between NK cell function and oral PFOS exposure in male mice. At 0.5 and 5 mg PFOS/kg TAD, PFOS caused increased NK cell activity, yet splenic NK cell activity was significantly decreased at a level of ≥ 50 mg PFOS/kg TAD. Peden-Adams et al. (Citation2008) reported that, when adult B6C3F1 mice were exposed to PFOS daily via gavage for 28 days (at 0, 0.005, 0.05, 0.1, 0.5, 1, or 5 mg PFOS/kg TAD), plasma lysozyme activity was only altered at the 0.1 and 5.0 mg/kg levels, and SRBC-specific IgM production was increased and then suppressed with increasing doses of PFOS. Based upon these types of outcomes in the above-cited studies, it is reasonable to state that the effects observed at the doses used in the present study might not necessarily be the same as that at even lower (or higher) doses. Future studies will need to be undertaken to verify this point.
In summary, the present study demonstrated that exposure to PFOS could result in generation of ROS, and may overwhelm the homeostasis of anti-oxidative systems. Activation of caspase-3 and -9, DNA laddering, and apoptosis were clearly evident. However, the underlying mechanisms of effects of these compounds on ROS generation and mitochondria permeability remain unknown. In addition, further investigation is needed to determine if the activation of caspases is mediated through cytochrome c-dependent apoptosis pathways and the release of cytochrome c.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81172630), and the Key Laboratory of Industrial Ecology and Environmental Engineering of China Ministry of Education (KLIEEE-10-01).
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Alexander, B. H., Olsen, G. W., Burris, J. M., Mandel, J. H.Mandel, J. S. 2003. Mortality of employees of a perfluorooctanesulphonyl fluoride manufacturing facility. Occup. Environ. Med. 60:722–729.
- Andersen, C. S., Fei, C., Gamborg, M., Nohr, E. A., Sørensen, T. I.Olsen, J. 2010. Prenatal exposures to perfluorinated chemicals and anthropometric measures in infancy. Am. J. Epidemiol. 172:1230–1237.
- Bai, J., Rodriguez, A. M., Melendez, J. A.Cederbaum, A. I. 1999. Over-expression of catalase in cytosolic or mitochondrial compartment protects HepG2 cells against oxidative injury. J. Biol. Chem. 274:26217–26224.
- Berger, J., Machackova, M., Berger, Z. 2005. Effects of feed restriction on the nucleolar structure and function in lymphocytes. Basic. Clin. Pharmacol. Toxicol 97:236–237.
- Brieger, A., Bienefeld, N., Hasan, R., Goerlich, R.Haase, H. 2011. Impact of perfluorooctane sulfonate and perfluorooctanoic acid on human peripheral leukocytes. Toxicol. In Vitro 25:960–968.
- Burris, J. M., Lundberg, J. K., Olsen, G. W., Simpson, C., and Mandel, J. H. 2002. Determination of Serum Half-lives of Several Fluorochemicals. Interim Report No. 2. St. Paul, MN: 3M Company. U.S. EPA docket AR-226–1086. Washington, DC: US Environmental Protection Agency.
- Chen, T., Zhang, L., Yue, J. Q., Lv, Z. Q., Xia, W., Wan, Y. J., Li, Y. Y.Xu, S. Q. 2011. Prenatal PFOS exposure induces oxidative stress and apoptosis in the lung of rat offspring. Reprod. Toxicol. DOI: 10.1016/j.reprotox.2011.03.003.
- Corsini, E., Sangiovanni, E., Avogadro, A., Galbiati, V., Viviani, B., Marinovich, M., Galli, C. L., Dell’agli, M.Germolec, D. R. 2012. In vitro characterization of the immunotoxic potential of several perfluorinated compounds (PFC). Toxicol. Appl. Pharmacol. 258:248–255.
- Darzynkiewicz, Z., Bruno, S., Del Bino, G., Gorczyca, W., Hotz, M. A., and Lassota, P. 1992. Features of apoptosis cells measured by flow cytometry. Cytometry 13:795–808.
- de Vos, M. G., Huijbregts, M. A., van den Heuvel-Greve, M. J., Vethaak, A. D., van de Vijver, K. I., Leonards, P. E., van Leeuwen, S. P., de Vooqt, P.Hendriks, A. J. 2008. Accumulation of perfluorooctane sulfonate (PFOS) in the food chain of the Western Scheldt estuary: Comparing field measurements with kinetic modeling. Chemosphere 70:1766–1773.
- Dewitt, J. C., Peden-Adams, M. M., Keller, J. M., andGermolec, D. R. 2012. Immunotoxicity of perfluorinated compounds: Recent developments. Toxicol. Pathol. 40:300–311.
- Dong, G. H., Zhang, Y. H., Zheng, L., Liu, W., Jin, Y. H.He, Q. C. 2009. Chronic effects of perfluorooctane sulfonate exposure on immunotoxicity in adult male C57BL/6 mice. Arch. Toxicol. 83:805–815.
- Egeghy, P. P.Lorber, M. 2011. An assessment of the exposure of Americans to perfluorooctane sulfonate: A comparison of estimated intake with values inferred from NHANES data. J. Exposure Sci. Environ. Epidemiol. 21:150–168.
- Elcombe, C. R., Elcombe, B. M., Foster, J. R., Chang, S. C., Ehresman, D. J., Noker, P. E., and Butenhoff, J. L. 2012. Evaluation of hepatic and thyroid responses in male Sprague Dawley rats for up to eighty-four days following seven days of dietary exposure to potassium perfluo-rooctanesulfonate. Toxicology 293:30–40.
- Fei, C., McLaughlin, J. K., Lipworth, L.Olsen, J. 2010. Prenatal exposure to PFOA and PFOS and risk of hospitalization for infectious diseases in early childhood. Environ. Res. 110:773–777.
- Francis, J., MohanKumar, S. M., and MohanKumar, P. S. 2000. Correlations of norepinephrine release in the paraventricular nucleus with plasma corticosterone and leptin after systemic lipopolysaccharide: blockade by soluble IL-1 receptor. Brain. Res. 867:180–187.
- Giesy, J. P., and Kannan, K. 2001. Global distribution of perfluorooctane sulfonate in wildlife. Environ. Sci. Technol. 35:1339–1342.
- Gilliland, F. D.Mandel, J. S. 1993. Mortality among employees of a perfluorooctanoic acid production plant. Occup. Med. 35:950–954.
- Hansen, K. J., Clemen, L. A., Ellefson, M. E., and Johnson, H. O. 2001. Compound-specific, quantitative characterization of organic fluorochemicals in biological matrices. Environ. Sci. Technol. 35:766–770.
- Hoffman, K., Webster, T. F., Weisskopf, M. G., Weinberg, J.Vieira, V. M. 2010. Exposure to polyfluoroalkyl chemicals and attention deficit hyperactivity disorder in U.S. children aged 12–15 years. Environ. Health. Perspect. 118:1762–1767.
- Hosea, H. J., Rector, E. S., and Taylor, C. G. 2004. Dietary repletion can replenish reduced T-cell subset numbers and lymphoid organ weight in zinc-deficient and energy-restricted rats. Br. J. Nutr. 91:741–747.
- Hu, X. Z.Hu, D. C. 2009. Effects of perfluuorooctanoate and perfluuorooctane sulfonate exposure on hepatoma Hep G2 cells. Arch. Toxicol. 83:851–861.
- Jin, Y., Saito, N., Harada, K. H., Inoue, K.Koizumi, A. 2007. Historical trends in human serum levels of perfluorooctanoate and perfluorooctane sulfonate in Shenyang, China. Tohoku J. Exp. Med. 212:63–70.
- Joensen, U. N., Bossi, R., Leffers, H., Jensen, A. A., Skakkebaek, N. E., and Jørgensen, N. 2009. Do perfluoroalkyl compounds impair human semen quality? Environ. Health Perspect. 117:923–927.
- Keil, D. E., Mehlmann, T., Butterworth, L., and Peden-Adams, M. M. 2008. Gestational exposure to perfluorooctane sulfonate suppresses immune function in B6C3F1 mice. Toxicol. Sci. 103:77–85.
- Kelly, S. A., Havrilla, C. M., Brady, T. C., Abramo, K. H., and Levin, E. D. 1998. Oxidative stress in toxicology: Established mammalian and emerging piscine model systems. Environ. Health Perspect. 106:375–384.
- Kim, H. S., Jun Kwack, S., Sik Han, E., Seok Kang, T., Hee Kim, S., and Young Han, S. 2011. Induction of apoptosis and CYP4A1 expression in Sprague-Dawley rats exposed to low doses of perfluorooctane sulfonate. J. Toxicol. Sci. 36:201–210.
- Langheinrich, U., Hennen, E., Stott, G., and Vacun, G. 2002. Zebrafish as a model organism for the identification and characterization of drugs and genes affecting p53 signaling. Curr. Biol. 12:2023–2028.
- Lau, C., Anitole, K., Hodes, C., Lai, D., Pfahles-Hutchens, A., and Seed, J. 2007. Perfluoroalkyl acids: A review of monitoring and toxicological findings. Toxicol. Sci. 99:366–394.
- Lefebvre, D. E., Curran, I., Armstrong, C., Coady, L., Parenteau, M., Liston, V., Barker, M., Aziz, S., Rutherford, K., Bellon-Gagnon, P., Shenton, J., Mehta, R.Bondy, G. 2008. Immunomodulatory effects of dietary potassium perfluorooctane sulfonate (PFOS) exposure in adult Sprague-Dawley rats. J. Toxicol. Environ. Health 71:1516–1525.
- Liu, C., Yu, K., Shi, X., Wang, J., Lam, P. K., Wu, R. S., and Zhou, B. 2007. Induction of oxidative stress and apoptosis by PFOS and PFOA in primary cultured hepatocytes of freshwater tilapia (Oreochromis niloticus). Aquat. Toxicol. 82:135–143.
- Livingstone, D. R. 2001. Contaminant-stimulated reactive oxygen species production and oxidative damage in aquatic organisms. Marine Pollut Bull 42:656–666.
- Luster, M. I.Rosenthal, G. J. 1993. Chemical agents and the immune response. Environ. Health. Perspect. 100:219–226.
- Melzer, D., Rice, N., Depledge, M. H., Henley, W. E.Galloway, T. S. 2010. Association between serum perfluoroctanoic acid (PFOA) and thyroid disease in the NHANES Study. Environ. Health. Perspect. 118:686–692.
- Meyer, J. N., Smith, J. D., Winston, G. W., and Di Giulio, R. T. 2003. Anti-oxidant defenses in killifish (Fundulus heteroclitus) exposed to contaminated sediments and model pro-oxidants: Short-term and heritable responses. Aquat. Toxicol. 65:377–395.
- Mollenhauer, M. A., Bradshaw, S. G., Fair, P. A., McGuinn, W. D., and Peden-Adams, M. M. 2011. Effects of perfluorooctane sulfonate (PFOS) exposure on markers of inflammation in female B6C3F1 mice. J. Environ. Sci. Health 46:97–108.
- Netto, L. E., Kowaltowski, A. J., Castilho, R. F., and Vercesi, A. E. 2002. Thiol enzymes protecting mitochondria against oxidative damage. Meth. Enzymol. 348:260–270.
- Noorlander, C. W., van Leeuwen, S. P., Te Biesebeek, J. D., Mengelers, M. J.Zeilmaker, M. J. 2011. Levels of perfluorinated compounds in food and dietary intake of PFOS and PFOA in the Netherlands. J. Agric. Food Chem. 59:7496–7505.
- O’Brien, T. M.Wallace, K. B. 2004. Mitochondrial permeability transition as the critical target of N-acetyl perfluorooctane sulfonamide. Toxicol. Sci. 82:333–340.
- OECD (Organization for Economic Co-operation and Development). 2002. Hazard Assessment of Perfluorooctane Sulfonate (PFOS) and Its Salts. Available online at: http://www.oecd.org/ dataoecd/ 23/18/2382880.pdf.
- Panaretakis, T., Shabalina, I. G., Grandér, D., Shoshan, M. C.DePierre, J. W. 2001. Reactive oxygen species and mitochondria mediate the induction of apoptosis in human hepatoma HepG2 Cells by the rodent peroxisome proliferator and hepatocarcinogen, perfluorooctanoic acid. Toxicol. Appl. Pharmacol. 173:56–64.
- Peden-Adams, M. M., EuDaly, J. G., Dabra, S., EuDaly, A., Heesemann, L., Smythe, J., and Keil, D. E. 2007. Suppression of humoral immunity following exposure to the perfluorinated insecticide sulfluramide. J. Toxicol. Environ. Health 70:1130–1141.
- Peden-Adams, M. M., Keller, J. M., and EuDaly, J. G. 2008. Suppression of humoral immunity in mice following exposure to perfluorooctane sulfonate. Toxicol. Sci. 104:144–154.
- Pulido, M. D.Parrish, A. R. 2003. Metal-induced apoptosis: Mechanisms. Mutat. Res. 533:227–241.
- Qazi, M. R., Bogdanska, J., Butenhoff, J. L., Nelson, B. D., DePierre, J. W., and Abedi-Valugerdim, M. 2009. High-dose, short-term exposure of mice to perfluorooctanesulfonate (PFOS) or perfluorooctanoate (PFOA) affects the number of circulating neutrophils differently, but enhances the inflammatory responses of macrophages to lipopolysaccharide (LPS) in a similar fashion. Toxicology 262:207–214.
- Santos, M. A., Pacheco, M.Ahmad, I. 2004. Anguilla anguilla L. anti-oxidants responses to in situ bleached kraft pulp mill effluent outlet exposure. Environ. Int. 30:301–308.
- Sedlak, M. D.Greig, D. J. 2012. Perfluoroalkyl compounds (PFCs) in wildlife from an urban estuary. J. Environ. Monit. 14:146–154.
- Sheikh, M. S.Fornace, A. J. Jr. 2000. Role of p53 family members in apoptosis. J. Cell. Physiol. 182:171–181.
- Stein, C. R., Savitz, D. A.Dougan, M. 2009. Serum levels of perfluorooctanoic acid and perfluorooctane sulfonate and pregnancy outcome. Am. J. Epidemiol. 170:837–846.
- Stockholm Convention on Persistent Organic Pollutants. Governments unite to step-up reduction on global DDT reliance and add nine new chemicals under international treaty. Geneva, Stockholm Convention Secretariat, 9 May 2009. Available online at: http://chm.pops.int/ Convention/Pressrelease/COP4Geneva8May2009/tabid/542/language/en-US/Default.aspx
- Vermes, I., Haanen, C., Steffens-Nakken, H., Reutelingsperger, C. 1995. A novel assay for apoptosis, flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein-labeled Annexin V. J. Immunol. Methods. 184:39–45.
- Wang, J. F., Jerrells, T. R.Spitzer, J. J. 1996. Decreased production of reactive oxygen intermediates is an early event during in vitro apoptosis of rat thymocytes. Free Rad. Biol. Med. 20:533–542.
- Wang, Y., Wang, L., Liang, Y., Qiu, W., Zhang, J., Zhou, Q.Jiang, G. 2011. Modulation of dietary fat on the toxicological effects in thymus and spleen in BALB/c mice exposed to perfluorooctane sulfonate. Toxicol. Lett. 204:174–182.
- Weller, I. 2001. Secondary immunodeficiency. In: Immunology. 6th Edition (Roitt, I., Brostoff, J.Male, D. K. , Eds.). Edinburgh, Scotland: Mosby, pp. 315–317.
- Zheng, L., Dong, G. H., Zhang, Y. H., Liang, Z. F., Jin, Y. H., and He, Q. C. 2011. Type 1 and type 2 cytokines imbalance in adult male C57Bl/6 mice following a 7-day oral exposure to perfluorooctanesulfonate (PFOS). J. Immunotoxicol. 8:30–38.