
Abstract
Exposure to subacute ozone (O3) causes pulmonary neutrophil recruitment. In mice, this recruitment requires IL-17A. Ozone also causes expression of IL-23 and IL-1, which can induce IL-17A. The purpose of this study was to examine the hypothesis that IL-23 and IL-1 contribute to IL-17A expression and subsequent neutrophil recruitment after subacute O3 exposure. Wild-type, IL-23−/−, and Flt3l−/− mice were exposed to air or 0.3 ppm O3 for 72 h. Flt3l−/− mice lack conventional dendritic cells (cDC) that can express IL-23 and IL-1. Other wild-type mice were pre-treated with saline or the IL-1R1 antagonist anakinra prior to O3 exposure. After exposure, bronchoalveolar lavage (BAL) was performed and lung tissue harvested. The results indicated that pulmonary Il17a mRNA abundance and IL-17A+ F4/80+ cells were significantly reduced in O3-exposed IL-23−/− vs in wild-type mice. In contrast, anakinra had no effect on Il23a or Il17a pulmonary mRNA abundance or on BAL concentrations of the neutrophil survival factor G-CSF, but anakinra did reduce BAL neutrophil numbers, likely because anakinra also reduced BAL IL-6. Compared to air, O3 caused a significant increase in DC numbers in wild-type, but not in Flt3−/− mice. However, there was no significant difference in Il23a or Il17a mRNA abundance or in BAL neutrophil count in O3-exposed Flt3−/− vs in wild-type mice. From these results, it was concluded that IL-23 but not IL-1 contributes to the IL-17A expression induced by subacute O3 exposure. Induction of IL-23 by O3 does not appear to require cDC.
Keywords:
Introduction
Exposure to the air pollutant, ozone (O3), causes asthma symptoms (Gent et al. Citation2003), increases susceptibility to respiratory infections (Stanek et al. Citation2011) and even increases the risk of mortality (Bell et al. Citation2004; Ito et al. Citation2005). These responses to O3 may be consequences of lung epithelial barrier disruption (Kehrl et al. Citation1987; Lang et al. Citation2008), impaired immune cell function within the respiratory tract (Gilmour et al. Citation1991, Citation1993) and recruitment of neutrophils (PMN) to the lungs (Alexis et al. Citation2013; Barreno et al. Citation2013; Kasahara et al. Citation2013).
This laboratory has previously reported that, in mice, IL-17A contributes to the PMN recruitment observed following subacute O3 exposure (0.3 ppm for 24–72 h), at least in part via effects of IL-17A on expression of G-CSF, a PMN survival factor (Kasahara et al. Citation2012; Mathews, Williams, et al. Citation2014). Sources of IL-17A in the lung after subacute O3 include macrophages and γδ T-cells (Kasahara et al. Citation2012; Mathews, Williams, et al. Citation2014). IL-23 can promote IL-17A expression in T-cells including γδ T-cells (Gaffen et al. Citation2014). The receptor for IL-23 is widely expressed on immune cells (Parham et al. Citation2002) and binding of IL-23 to its receptor induces the activation of multiple transcriptional pathways that lead to the expression of IL-17A (Harrington et al. Citation2006). IL-23 expression is induced in the lungs following subacute O3 exposure (Mathews, Williams, et al. Citation2014), but its role in subsequent IL-17A expression and PMN recruitment has not been established. IL-1 also has the capacity to increase IL-17A expression in the lungs (Chung et al. Citation2009; Gasse et al. Citation2011; Kim et al. Citation2014), perhaps because IL-1 can induce IL-23 expression (Harris et al. Citation2008; Shainheit et al. Citation2008). Both IL-1α and IL-1β are induced in the lungs following subacute O3 exposure and IL-1 contributes to the subsequent PMN recruitment induced by O3 (Johnston et al. Citation2007), but whether IL-1-induced changes in IL-17A expression, either directly or via changes in IL-23, contribute to these effects on PMN recruitment has not been established.
The present study was done to validate the hypothesis that IL-23 and IL-1 contribute to the production of IL-17A after subacute O3 exposure. To address this hypothesis, IL-23−/− and wildtype (WT) control counterparts were exposed to O3 (0.3 ppm O3 for 72 h) and ensuing pulmonary inflammation was measured. WT mice were also treated with the IL-1R antagonist anakinra or saline prior to O3 exposure to determine whether effects of IL-1 were mediated via an ability to induce IL-23 and/or IL-17A expression.
Dendritic cells (DC) are among the sources of IL-23 in the lungs (Aggarwal et al. Citation2003; Zhang et al. Citation2015). While the role of DC as antigen-presenting cells is perhaps more widely appreciated, DC also participate in innate immune responses. DC express toll-like receptors, nod-like receptors and the CD1 family of receptors, enabling DC to quickly respond to pathogen- and damage-associated molecular patterns (DAMPs). DAMPs such as hyaluronan, nucleosides and the high-mobility box group 1 protein are produced in the lungs following O3 exposure (Hollingsworth et al. Citation2007) and the number of DC in the lungs increases after O3 exposure (Brand et al. Citation2012). Whether DC contribute to the IL-23 and IL-1 induced following O3 has not been established. However, the observations that DC lie directly adjacent to γδ T-cells in the lungs (Wands et al. Citation2005) and that γδ T-cells are important sources of IL-17A required for O3-induced PMN recruitment (Mathews, Williams, et al. Citation2014) suggest DC may indeed be involved.
Consequently, the present study also examined the hypothesis that DC are an important source of the IL-23 driving IL-17A expression after subacute O3 exposure. To address this hypothesis, fms-like tyrosine kinase-3 ligand null (Flt3l−/−) mice and their WT controls were exposed to O3 (0.3 ppm O3 for 72 h) or air and ensuing pulmonary inflammation was measured. Flt3l is required for differentiation of the common myeloid progenitor of cDC (Waskow et al. Citation2008). Thus, Flt3l−/− mice lack conventional DC (cDC) that represent the majority of lung DC (Hammad and Lambrecht Citation2011).
Materials and methods
Animals
IL-23−/− mice on a C57BL/6 background (Ghilardi et al. Citation2004) were kindly provided by Nico Ghilardi (Genentech, San Francisco, CA). IL-23−/− and littermate control wild-type (WT) mice were bred from IL-23+/− breeding pairs. IL-23−/− mice display no overt phenotype. Development, fertility, size and weight are all normal and in the absence of stimulation there is no apparent effect on immune system development aside from a small increase in CD8+ DC (Ghilardi et al. Citation2004). Flt3l−/− mice (McKenna et al. Citation2000) and gender- and age-matched WT (C57BL/6) control mice were purchased from Taconic Biosciences (Hudson, NY). For other experiments, including experiments involving anakinra treatment, WT (C57BL/6) mice were purchased from The Jackson Laboratory (Jackson Labs, Bar Harbor, ME). All mice were provided ad libitum access to a standard mouse chow diet and filtered tap water. All mice were 8–12-weeks-old at the time of study and were acclimated for 1 week before exposure. All mice were housed in a vivarium maintained at 21–22 °C with a 48–50% relative humidity and a 12-h light:dark cycle. The Harvard Medical Area Standing Committee on Animals approved all aspects of this study.
Protocol
To study the role of IL-23 in responses to O3, WT and IL-23−/− mice were exposed to 0.3 ppm O3 for 72 h, as previously described (Kasahara et al. Citation2012). Following exposure, mice were immediately euthanized by overdose with an intraperitoneal injection of sodium pentobarbital. The trachea was then exposed, canulated and bronchoalveolar lavage (BAL) was performed in situ. The lungs were then cleared of blood by perfusing the right ventricle with 10 ml of ice-cold phosphate-buffered saline (PBS, pH 7.4) after creating an incision in the left ventricle. The right lung was used for flow cytometry and the left lung lobes were placed in RNAlater (Qiagen, Germantown, MD) for subsequent preparation of RNA for real time PCR.
To study the role of IL-1α and IL-1β, a different set of WT mice were treated with a recombinant form of human IL-1 receptor antagonist anakinra or with saline. Anakinra is efficacious in mice (Alexander et al. Citation1991) and both IL-1α and IL-1β require IL-1R1 for signaling (So et al. Citation2007). Anakinra (100 mg/kg, intraperitoneally, Swedish Orphan Biovitrium AB, Stockholm, Sweden) or an equal volume of saline was administered 24 h before, immediately prior to and every 24 h during the O3 exposure protocol for a total of four injections. The dose of anakinra used here was based on prior studies such as Alexander et al. (Citation1991). These animals were euthanized immediately after O3 exposure and BAL and lung tissue collection performed as above. A similar protocol to that used for IL-23−/− mice and their controls was used to examine the role of cDC, using Flt3l−/− and their WT controls.
Bronchoalveolar (BAL) lavage
BAL was performed on each mouse by instilling two aliquots of 1 ml ice-cold PBS into the lung via the trachea. After centrifugation, BAL supernatant was stored at −80 °C until assayed. BAL cells were centrifuged onto glass slides and stained using the Hema3 Stain kit (equivalent to Wright-Geimsa). At least 300 cells were counted. BAL cytokines and chemokines were measured using a multiplex assay (Eve Technologies, Calgary, Alberta, Canada). Total BAL protein was measured using a Bradford assay (Bio-Rad, Hercules, CA).
Flow cytometry
The left lung of each mouse was harvested and placed on ice in RPMI 1640 media containing 5% fetal bovine serum (FBS) and 20 mM HEPES (Life Technologies, Grand Island, NY). The lungs were then minced, returned to the media and incubated at 37 °C with 2.5 mg/ml collagenase IV (Roche, Mannheim, Germany) for 30 min while rotating. Cells were then passed through a 20-gauge needle and a 70-μm mesh. Non-specific Fc receptor binding was blocked (TruStain fcX; Biolegend, San Diego, CA) and the cells were then stained for flow cytometry. The conjugated antibodies allophycocyanin (APC)-anti-CD11c (clone: N418), fluorescein isothiocyanate (FITC)- and AlexaFluor (AF)-647-anti-MHC-II (clone: M5/114.15.2) and phycoerythrin (PE)-anti-Siglec-F (clone: E50-2440) (all BD Biosciences, San Jose, CA) were used to identify cDC and macrophages; PE-anti-TCRδ (clone: GL3), PE-Cγ7-anti-CD45 (clone: 30-F11) and FITC-anti-CD3 (clone: 17A2) (all Biolegend) were used to identify γδ T-cells.
To investigate IL-17A production by γδ T-cells, single cell lung suspensions were incubated with or without PMA (phorbol 12-myristate 13-acetate; 100 ng/ml), ionomycin (500 ng/ml) and GolgiStop (BD Biosciences) for 4 h prior to staining. Intracellular IL-17A was detected after permeablization (Cytofix/Cytoperm; BD Biosciences) and staining with APC-anti-IL-17A within γδ T-cells or biotinylated-anti-IL-17A (clone: TC11-18H10.1) with a streptavidin-PE secondary antibody within macrophages. Isotype control antibodies were used to set all gates. Cells were analyzed using a Canto II (BD Biosciences) and the data was analyzed using FlowJo software (Tree Star; Ashland, OR). A minimum of 40 000 events was acquired for each sample.
Quantitative real-time PCR
The right lung was homogenized and total RNA purified using a RNeasy Column kit (Qiagen, Germantown, MD). RNA quantity and purity was determined using a small volume spectrophotometer (Nanodrop; Thermo Scientific, Waltham, MA). A total of 1 μg RNA was converted into cDNA using a Super Script III First-strand amplification kit for qRT-PCR (Life Technologies, Grand Island, NY). Il17a and Il23a mRNA abundances were quantitated by real-time PCR (7300 Real-Time PCR Systems; Applied Biosystems, Carlsbad, CA) using intron-spanning primers (Kasahara et al. Citation2012; Mathews, Joel, et al. Citation2014) and SYBR-green detection. Il17a and Il23a were normalized to 36b4 expression using ΔΔCt method (Livak and Schmittgen Citation2001).
Statistical analysis
Data were analyzed by one-way analysis of variance (ANOVA) followed by a post-hoc Tukey’s t-test, linear regression (Prism 5.0; GraphPad Software, La Jolla, CA) or factorial ANOVA with a post-hoc Fisher-least significant difference test (Statistica; StatSoft, Tulsa, OK). Non-normally distributed data was log transformed prior to statistical analysis. A p-value < 0.05 was considered statistically significant.
Results
IL-23 deficiency reduces O3-induced IL-17A expression
Compared to air, O3 (0.3 ppm for 72 h) caused a significant increase in both pulmonary Il23a and Il17a mRNA abundances (), as noted earlier. To determine whether IL-23 contributed to induction of Il17a expression, pulmonary Il17a mRNA abundance (BAL IL-17A was below limit of detection of ELISA) was measured in WT and IL-23−/− mice exposed to O3. Il17a mRNA abundance was significantly reduced in IL-23−/− vs in WT mice ().
Figure 1. (A) Pulmonary Il23a and (B) Il17a mRNA abundance, determined by qRT-PCR, in WT mice exposed to room air or ozone (0.3 ppm) for 72 h. Results shown are means ± SE from 5–6 mice/group and are expressed relative to the air exposed mice. *p < 0.05 vs air.
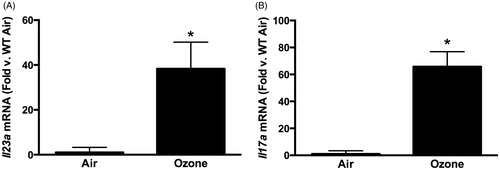
Figure 2. (A) Pulmonary Il17a mRNA abundance (expressed relative to in WT mice). (B) Pulmonary IL-17A+ γδ T-cells. (C) Pulmonary IL-17A+ interstitial macrophages measured by flow cytometry in WT and IL-23−/− mice exposed to ozone (0.3 ppm for 72 h). Results shown are means ± SEM of 8–9 mice/group for qRT-PCR and four mice/group for flow cytometry. *p < 0.05 vs WT mice.
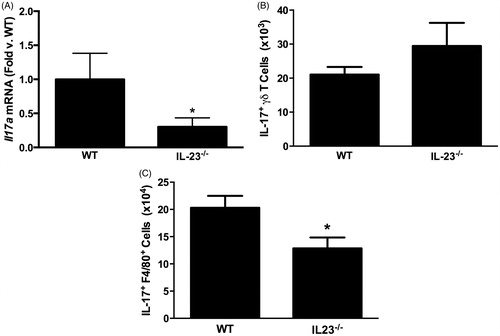
Earlier studies in this laboratory noted that both interstitial macrophages and γδ T-cells express IL-17A after O3 exposure. To determine which cell type was impacted by an IL-23 deficiency, flow cytometry was used to quantify IL-17A expressing macrophages and γδ T-cells in the lungs of WT and IL-23−/− mice exposed to O3. The total number of γδ T-cells was not different in O3-exposed WT and IL-23−/− (data not shown), nor was there any difference in the number of IL-17A+ γδ T-cells (). In contrast, IL-17A+ macrophages () were lower in the lungs of the IL-23−/− vs in those of the WT mice.
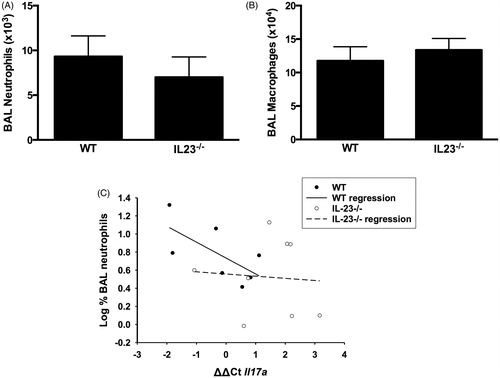
Despite the significant reduction in Il17a mRNA expression in IL-23−/− vs in WT mice () and data indicating that IL-17A is required for PMN recruitment after subacute O3 exposure (Kasahara et al. Citation2012; Mathews et al. Citation2013), we observed only a non-significant (≈25%) reduction in BAL PMN in O3-exposed IL-23−/− vs WT mice (), nor was there any change in BAL macrophage numbers (). Similarly, IL-23 deficiency resulted in a reduction in BAL G-CSF concentrations (86 [± 40] vs 52 [± 12] pg/ml in WT and IL-23−/− mice, respectively), but the effect was not significant. There were also no significant IL-23-dependent changes in BAL concentrations of other cytokines that either are involved (IL-6, CXCL5, CXCL1, CXCL2, IL-1) or might be involved (CCL3, CCL5) in neutrophil recruitment to the lungs after O3 exposure (data not shown). Nevertheless, IL17a mRNA expression did appear to account for at least part of the variance in BAL PMN among WT mice, since there was a significant inverse correlation (r2 = 0.444, p < 0.05) between the percentage of BAL cells that were PMN and Il17a mRNA abundance () in WT mice (note that an increase in ΔΔCt indicated reduced Il17a expression). In contrast, no such correlation existed in the IL-23−/− mice (r2 = 0.005, NS) ().
Figure 3. Bronchoalveolar lavage (BAL) (A) neutrophils and (B) macrophages in WT and IL-23−/− mice exposed to ozone (0.3 ppm for 72 h). Results shown are means ± SE of 8–9 mice/group. (C) Log %BAL neutrophils plotted vs ΔΔCt values for Il17a. All mice were ozone-exposed. The regression line shown was calculated from the combined data from the WT and IL-23−/− mice. Note: increase in ΔΔCt indicates reduced Il17a expression.
IL-1 contributes to O3-induced PMN recruitment via an IL-17A-/IL-23-independent mechanism
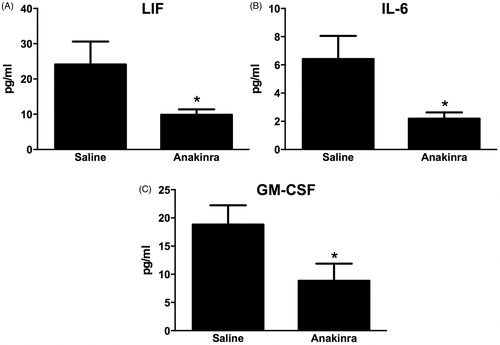
Compared to saline, there was no effect of the IL-1R1 antagonist anakinra on either Il17a or Il23 expression in O3-exposed mice (). Nevertheless, O3-induced increases in BAL PMN numbers were significantly lower in anakinra- vs saline-treated mice (), although macrophage numbers and BAL protein concentrations (a measure of O3-induced lung injury) were not affected (). A multiplex assay was used to measure other IL-1-dependent cytokines and chemokines that could be contributing to the changes in PMN numbers. The data shown in illustrate that IL-6, LIF and GM-CSF concentrations were each significantly reduced in anakinra-treated vs in saline-treated mice exposed to O3.
Figure 4. Pulmonary (A) Il17a and (B) Il23a mRNA abundance (expressed relative to WT mice) and BAL (C) neutrophils, (D) macrophages and (E) total protein in WT mice exposed to ozone (0.3 ppm for 72 h) that had been treated with anakinra or saline. Results shown are means ± SE of 6–12 mice/group. *p < 0.05 vs saline-treated mice.
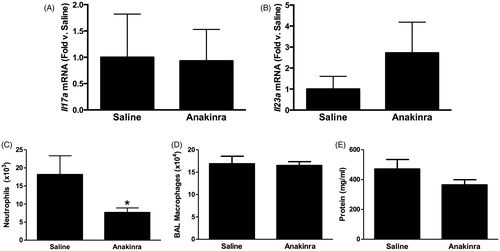
Figure 5. BAL (A) LIF, (B) IL-6 and (C) GM-CSF assayed by multiplex from WT (C57BL/6J) mice exposed to ozone (0.3 ppm for 72 h) that had been treated with anakinra or saline. Results shown are means ± SE of eight mice/group. * p < 0.05 vs saline-treated mice.
Conventional dendritic cells do not contribute to O3-induced PMN recruitment to the lungs
Although other O3 exposure paradigms have been shown to increase pulmonary DC numbers (Brand et al. Citation2012), the effects of subacute O3 (0.3 ppm for 24–72 h) have not been established. Consequently, flow cytometry was used here to quantify the number of DC in lungs of mice exposed to room air or to O3 for 24, 48 or 72 h. DC (CD11c+MHCII+Siglec-F− cells) (Desch et al. Citation2014) were identified using the gating strategy shown in . In WT mice, O3 caused a time-dependent increase in the number of pulmonary DC (): DC counts were not different in air-exposed mice and mice exposed to O3 for 24 h, but O3 exposure for 48 or 72 h did increase the number of pulmonary DC.
Figure 6. (A) Gating strategy used to assess lung DC. Cells were first gated based on forward scatter (FSC) and side-scatter (SSC) characteristics. Cells positive for MCH-II and CD11c were determined to be either dendritic cells (DC, SiglecF−) or macrophages M∅, SiglecF+). (B) Total number of MHC-II+/CD11c+/SiglecF− DC in WT mice exposed to air or ozone (0.3 ppm) for 24, 48 or 72 h. (C) Total number of MHC-II+/CD11c+/SiglecF− DC in WT and Flt3l−/− mice exposed to air or ozone (0.3 ppm) for 72 h. Results shown are means ± SE of 7–8 mice/group. *p < 0.05 vs air-exposed mice with same genotype; #p < 0.05 vs WT mice with same exposure.
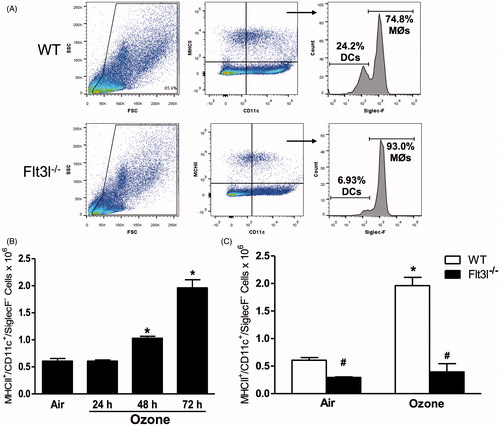
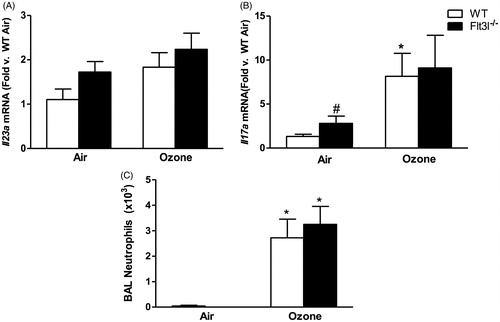
To determine whether cDC contributed to pulmonary Il23a and Il17a expression after O3 exposure, Flt3l−/− mice which lack cDCs due to a requirement for Flt3l in differentiation of the common myeloid progenitor (Waskow et al. Citation2008) were utilized. Indeed, DC were significantly lower in the lungs of Flt3l−/− than in WT mice and there was no O3-induced increase in DC numbers in the Flt3l−/− mice (). There was no difference in pulmonary Il23a or Il17a mRNA abundance in O3-exposed Flt3l−/− vs in WT mice (), although there was significantly more Il17a in the lungs of Flt3l−/− vs in WT mice exposed to air. The O3-induced increases in BAL PMN numbers were also unaffected by Flt3l genotype ().
Figure 7. Pulmonary (A) Il23a and (B) Il17a mRNA abundance (expressed relative to WT air mice) and (C) BAL neutrophil numbers in WT and Flt3l−/− mice exposed to room air or ozone (0.3 ppm) for 72 h. Results shown are means ± SE of 7–8 mice/group. *p < 0.05 vs air-exposed mice with same genotype; #p < 0.05 vs WT mice with same exposure.
Discussion
The data here indicate that IL-23 contributes to the IL-17A expression induced by subacute O3 exposure, at least in part via effects on lung interstitial macrophages. The data also indicate that IL-1 signaling contributes to the PMN recruitment induced by subacute O3 in an IL-17A- and IL-23-independent manner. Finally, the data demonstrate that, despite a robust increase in cDC numbers after O3 exposure, and the ability of these cells to express IL-23 in response to other stimuli (Fitch et al. Citation2007), cDCs are not the source of IL-23-dependent Il17a expression.
In O3-exposed mice, IL-23 deficiency caused a significant reduction in pulmonary Il17a mRNA abundance. These data were consistent with observations that Il23a expression is induced by subacute O3 exposure (Mathews, Williams, et al. Citation2014) and with data of other investigators showing that IL-23 promotes pulmonary IL-17A expression induced by other stimuli, including infection with Streptococcus pneumonia or Pseudomonas aeruginosa (Dubin et al. Citation2012; Kim et al. Citation2013). However, while Il17a mRNA was substantially reduced (≈70% reduction) in IL-23−/− vs WT mice (), it was not completely abolished. Similarly, Ghilardi et al. (Citation2004) observed only a partial reduction in T-cell IL-17 expression after in vitro stimulation. The results indicated that, while IL-23 promoted pulmonary IL-17A production after subacute O3 exposure, other factors that drive IL-17A were also important. For example, serum amyloid A, TGFβ, IL-6, IL-1 (Kim et al. Citation2013) and tumor necrosis factor (TNF)-α (Mathews, Williams, et al. Citation2014) can each induce IL-17A in various cell types. We previously reported no effect of IL-6 deficiency on IL-17A expression in WT mice after subacute O3 exposure, although IL-6 does contribute in adiponectin-deficient mice in which O3 induced much higher concentrations of IL-6 (Kasahara et al. Citation2014). (See additional discussion of other IL-17A promoting cytokines below.)
Flow cytometry was performed to determine which cells capable of expressing IL-17A were responsible for the IL-23 dependent changes in Il17a mRNA (). Subacute O3 exposure increases the numbers of IL-17A+ CD11c− macrophages and IL-17A+ γδ T-cells in the lungs (Kasahara et al. Citation2012; Mathews, Williams, et al. Citation2014). Both cell types express the IL-23R and the IL-12RB1 proteins necessary for responses to IL-23 (Parham et al. Citation2002). IL-23 can induce IL-17A expression in γδ T-cells (Lockhart et al. Citation2006; Sutton et al. Citation2009) in some settings. However, in a mouse model of colitis, IL-23 is not required for production of IL-17A from colonic γδ T-cells (Lee et al. Citation2015). Similarly, no reductions in IL-17+ γδ T-cells were observed in IL-23−/− mice exposed to O3, despite substantial reductions in Il17a mRNA expression in these mice. Instead, TNFα appears to contribute to expression of IL-17A in γδ T-cells after subacute O3.
IL-17A+ γδ T-cells were reduced ≈ 80% in TNFR2−/− vs in WT mice and the TNFα antagonist, etanercept, also caused substantial reductions in Il17a mRNA abundance in WT mice after subacute O3 exposure (Mathews, Williams, et al. Citation2014). In contrast, IL-17A+ macrophages were reduced in IL-23−/− mice. Similarly, IL-23 has been shown to induce IL-17A release from peripheral blood mononuclear cells from patients with atherosclerosis (Abbas et al. Citation2015). Macrophages also have the capacity to produce IL-23 (Bosmann et al. Citation2013; Abbas et al. Citation2015), suggesting these cells may use autocrine IL-23 signaling to promote IL-17A production. However, even IL-17A+ macrophages were reduced by only 50% in IL-23−/− mice and IL-17+ γδ T cells were unaffected, despite ∼70% reductions in Il17a mRNA abundance in these mice (). The results suggest that other unexamined cells may also contribute to IL-17A expression after O3 exposure. We have previously reported that TH17 cells are not involved (Kasahara et al. Citation2012), but innate lymphoid cells type 3 (ILC3) that also produce IL-17A are found in the lung in another condition of oxidative stress, i.e. obesity (Kim et al. Citation2014) and could be contributing to O3-induced increases in Il17a expression.
We have previously reported that, compared to isotype control antibody, anti-IL-17A causes a substantial reduction in BAL PMN numbers after subacute O3 exposure in mice (Mathews, Williams, et al. Citation2014). Hence, it was somewhat surprising that, despite ≈70% reduction in Il17a mRNA abundance in IL-23−/− vs in WT mice exposed to O3, there was no significant reduction in BAL PMN counts. The lack of effect of IL-23 deficiency on G-CSF and other cytokines and chemokines known to be required for O3-induced PMN recruitment (e.g. IL-6 and CXCL1) is consistent with the lack of effect of IL-23 deficiency on BAL PMN. Given the very marked increase in l17a mRNA abundance with O3 (≈60-fold) seen here, even with a 70% reduction in Il17a expression induced by IL-23 deficiency there would still be substantial Il17a expression in the IL-23−/− mice, which may be why PMN were only minimally affected. Additionally, increases in pulmonary Il23a mRNA abundance were not observed until 48 h of O3 exposure (Mathews, Williams, et al. Citation2014). In contrast, increases in pulmonary Il17a mRNA abundance were already observed after only 24 h of exposure, as were increases in IL-17A+ cell numbers, although Il17a continued to increase up to 72 h of exposure. Recruitment of BAL PMN to the lung also begins as early as 24 h after initiation of exposure (Mathews, Williams, et al. Citation2014). Taken together, the data here suggest to us that, after initiation of O3 exposure, factors other than IL-23 contribute to the early induction of IL-17A (likely from γδ T-cells) and consequent PMN recruitment, and that IL-23 induces additional IL-17A expression, likely from macrophages, only later in the exposure.
IL-1α and IL-1β are both expressed in the lungs after subacute O3 exposure (Johnston et al. Citation2007). Furthermore, IL-1R1 signaling has been shown to induce IL-23 expression in myeloid cells in response to other stimuli (Andersson et al. Citation2004). IL-1R1 signaling can also induce IL-17A expression in a variety of cell types (Chung et al. Citation2009; Gasse et al. Citation2011; Kim et al. Citation2014). Nevertheless, in the present study, there was no effect of the IL-1R1 antagonist anakinra on O3-induced increases in either pulmonary Il23a or Il17a mRNA abundance. However, given that anakinra was only administered 24 h prior to the initiation of O3 exposure, we cannot rule out the possibility that more sustained IL-1 inhibition might have been effective. In this context, it is conceivable the reduced Il17a mRNA abundance observed in the O3-exposed IL-23−/− mice might have been the result of developmental effects of IL-23 that promoted the ability of macrophages and other cells to subsequently produce IL-17A.
Whereas anakinra did not affect either Il17a or Il23a abundance in O3 exposed mice, it caused a substantial reduction in BAL PMN counts. The reduction in BAL PMN counts was consistent with previous observations of reduced BAL PMN numbers in IL-1R1−/− vs in WT mice exposed to subacute O3 (Johnston et al. Citation2007) and was likely a consequence of anakinra-induced reductions in BAL concentrations of IL-6, LIF and GM-CSF (). Both IL-6 and LIF can induce PMN recruitment and activation through binding of gp130 to their respective receptors (Murakami et al. Citation1993). Indeed, we previously reported reduced BAL PMN levels in IL-6−/− vs in WT mice after subacute O3 exposure (Johnston et al. Citation2005; Lang et al. Citation2008; Kasahara et al. Citation2014). GM-CSF also has the capacity to regulate chemotaxis and survival of PMN (Laan et al. Citation2003; Khajah et al. Citation2011) and IL-1β can induce the production of GM-CSF within the lung (Lukens et al. Citation2012).cDC have the capacity to release IL-23 during inflammation induced by a variety of stimuli, including cytokines, DAMPs and microbes (reviewed by Lambrecht and Hammad Citation2009). Nevertheless, no significant reduction in Il23a or Il17a mRNA abundance in Flt3l−/− vs in WT mice was observed here despite major reductions in the number of pulmonary DC in the Flt3l−/− mice. BAL PMN counts were also no different in Flt3l−/− vs in WT mice. Flt3l−/− mice are known to have a reduced white blood cell count, but this difference is largely attributed to a reduction in circulating lymphocytes rather than PMN (McKenna et al. Citation2000). The results indicated that subacute O3 exposure did not provide the stimuli necessary for induction of IL-23 in cDC. Indeed, studies in which IL-23 has been observed in cDC typically used antigenic stimulation (e.g. bacteria, autoantibodies) (Roses et al. Citation2008; Sutton et al. Citation2009). Instead, the IL-23 produced after subacute O3 exposure here likely originated from other myeloid cells rather than from cDC.
Conclusion
The data from the present study indicate that IL-23 is required, at least in part, for increases in pulmonary IL-17A expression that occur after subacute O3 exposure in mice and that one target of IL-23 is likely IL-17A+ interstitial macrophages. The data also indicate that O3-induced increases in pulmonary expression of IL-17A and IL-23 do not require IL-1, but that IL-1 nevertheless contributes to O3-induced neutrophil recruitment to the lungs, likely via effects on IL-6, LIF, and GM-CSF. Finally, this study showed that, despite increases in pulmonary cDC after subacute O3, these cells do not contribute to the IL-17A and IL-23 expression or the neutrophil recruitment induced by O3.
Acknowledgments
The authors would like to thank Nico Ghilardi (Genentech, San Francisco, CA) for providing IL-23−/− breeding pairs. This study was supported by ES013307 and ES000002. JAM was supported by ES02256. JDB was supported by HL007118.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Abbas A, Gregersen I, Holm S, Daissormont I, Bjerkeli V, Krohg-Sorensen K, Skagen KR, Dahl TB, Russell D, Almas T, et al. 2015. Interleukin 23 levels are increased in carotid atherosclerosis: Possible role for the IL-23/IL-17 axis. Stroke. 46:793–799.
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. 2003. IL-23 promotes a distinct CD4 T-cell activation state characterized by the production of IL-17. J Biol Chem. 278:1910–1914.
- Alexander HR, Doherty GM, Buresh CM, Venzon DJ, Norton JA. 1991. A recombinant human receptor antagonist to IL-1 improves survival after lethal endotoxemia in mice. J Exp Med. 173:1029–1032.
- Alexis NE, Zhou H, Lay JC, Harris B, Hernandez ML, Lu TS, Bromberg PA, Diaz-Sanchez D, Devlin RB, Kleeberger SR, et al. 2013. The glutathione-S-transferase mu 1 (GSTM1) null genotype and increased neutrophil response to low-level ozone (0.06 ppm). J Allergy Clin Immunol 131:610–612.
- Andersson A, Kokkola R, Wefer J, Erlandsson-Harris H, Harris RA. 2004. Differential macrophage expression of IL-12 and IL-23 upon innate immune activation defines rat auto-immune susceptibility. J Leukocyte Biol. 76:1118–1124.
- Barreno RX, Richards JB, Schneider DJ, Cromar KR, Nadas AJ, Hernandez CB, Hallberg LM, Price RE, Hashmi SS, Blackburn MR, et al. 2013. Endogenous osteopontin promotes ozone-induced neutrophil recruitment to the lungs and airway hyper-responsiveness to methacholine. Am J Physiol. 305:L118–129.
- Bell ML, McDermott A, Zeger SL, Samet JM, Dominici F. 2004. Ozone and short-term mortality in 95 US urban communities, 1987-2000. JAMA. 292:2372–2378.
- Bosmann M, Grailer JJ, Russkamp NF, Ruemmler R, Zetoune FS, Sarma JV, Ward PA. 2013. CD11c + alveolar macrophages are a source of IL-23 during lipopolysaccharide-induced acute lung injury. Shock. 39:447–452.
- Brand JD, Ballinger CA, Tuggle KL, Fanucchi MV, Schwiebert LM, Postlethwait EM. 2012. Site-specific dynamics of CD11b+ and CD103+ dendritic cell accumulations following ozone exposure. Am J Physiol. 303:L1079–L1086.
- Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, et al. 2009. Critical regulation of early TH17 cell differentiation by IL-1 signaling. Immunity. 30:576–587.
- Desch AN, Gibbings SL, Clambey ET, Janssen WJ, Slansky JE, Kedl RM, Henson PM, Jakubzick C. 2014. Dendritic cell subsets require cisactivation for cytotoxic CD8 T-cell induction. Nat Commun. 5:4674–4674.
- Dubin PJ, Martz A, Eisenstatt JR, Fox MD, Logar A, Kolls JK. 2012. IL-23-mediated inflammation in Pseudomonas aeruginosa pulmonary infection. Infect Immun. 80:398–409.
- Fitch E, Harper E, Skorcheva I, Kurtz SE, Blauvelt A. 2007. Pathophysiology of psoriasis: Recent advances on IL-23 and TH17 cytokines. Curr Rheumatol Rep. 9:461–467.
- Gaffen SL, Jain R, Garg AV, Cua DJ. 2014. IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat Rev Immunol. 14:585–600.
- Gasse P, Riteau N, Vacher R, Michel ML, Fautrel A, di Padova F, Fick L, Charron S, Lagente V, Eberl G, et al. 2011. IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PLoS One. 6:e23185.
- Gent JF, Triche EW, Holford TR, Belanger K, Bracken MB, Beckett WS, Leaderer BP. 2003. Association of low-level ozone and fine particles with respiratory symptoms in children with asthma. JAMA. 290:1859–1867.
- Ghilardi N, Kljavin N, Chen Q, Lucas S, Gurney AL, de Sauvage FJ. 2004. Compromised humoral and delayed-type hypersensitivity responses in IL-23-deficient mice. J Immunol. 172:2827–2833.
- Gilmour MI, Hmieleski RR, Stafford EA, Jakab GJ. 1991. Suppression and recovery of the alveolar macrophage phagocytic system during continuous exposure to 0.5 ppm ozone. Exp Lung Res. 17:547–558.
- Gilmour MI, Park P, Selgrade MK. 1993. Ozone-enhanced pulmonary infection with Streptococcus zooepidemicus in mice. The role of alveolar macrophage function and capsular virulence factors. Am Rev Respir Dis. 147:753–760.
- Hammad H, Lambrecht BN. 2011. Dendritic cells and airway epithelial cells at the interface between innate and adaptive immune responses. Allergy. 66:579–587.
- Harrington LE, Mangan PR, Weaver CT. 2006. Expanding the effector CD4 T-cell repertoire: the TH17 lineage. Curr Opin Immunol. 18:349–356.
- Harris KM, Fasano A, Mann DL. 2008. Cutting edge: IL-1 controls the IL-23 response induced by gliadin, the etiologic agent in celiac disease. J Immunol. 181:4457–4460.
- Hollingsworth JW, Kleeberger SR, Foster WM. 2007. Ozone and pulmonary innate immunity. Proc Am Thorac Soc. 4:240–246.
- Ito K, de Leon SF, Lippmann M. 2005. Associations between ozone and daily mortality: Analysis and meta-analysis. Epidemiology. 16:446–457.
- Johnston RA, Mizgerd JP, Flynt L, Quinton LJ, Williams ES, Shore SA. 2007. Type I IL-1 receptor is required for pulmonary responses to subacute ozone exposure in mice. Am J Respir Cell Mol Biol. 37:477–484.
- Johnston RA, Schwartzman IN, Flynt L, Shore SA. 2005. Role of IL-6 in murine airway responses to ozone. Am J Physiol. 288:L390–L397.
- Kasahara DI, Kim HY, Mathews JA, Verbout NG, Williams AS, Wurmbrand AP, Ninin FM, Neto FL, Benedito LA, Hug C, et al. 2014. Pivotal role of IL-6 in the hyper-inflammatory responses to subacute ozone in adiponectin-deficient mice. Am J Physiol. 306: L508–L520.
- Kasahara DI, Kim HY, Williams AS, Verbout NG, Tran J, Si H, Wurmbrand AP, Jastrab J, Hug C, Umetsu DT, et al. 2012. Pulmonary inflammation induced by subacute ozone is augmented in adiponectin-deficient mice: Role of IL-17A. J Immunol. 188:4558–4567.
- Kasahara DI, Williams AS, Benedito LA, Ranscht B, Kobzik L, Hug C, Shore SA. 2013. Role of the Adiponectin binding protein, T–cadherin (cdh13), in pulmonary responses to subacute ozone. PLoS One. 8:e65829.
- Kehrl HR, Vincent LM, Kowalsky RJ, Horstman DH, O'Neil JJ, McCartney WH, Bromberg PA. 1987. Ozone exposure increases respiratory epithelial permeability in humans. Am Rev Respir Dis. 135:1124–1128.
- Khajah M, Millen B, Cara DC, Waterhouse C, McCafferty DM. 2011. Granulocyte-macrophage colony-stimulating factor (GM-CSF): Chemoattractive agent for murine leukocytes in vivo. J Leukocyte Biol. 89:945–953.
- Kim BJ, Lee S, Berg RE, Simecka JW, Jones HP. 2013. IL-23 deficiency disrupts TH17 and TH1-related defenses against Streptococcus pneumoniae infection. Cytokine. 64:375–381.
- Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, et al. 2014. IL-17-producing innate lymphoid cells and NLRP3 inflammasome facilitate obesity-associated airway hyper-reactivity. Nat Med. 20:54–61.
- Laan M, Prause O, Miyamoto M, Sjöstrand M, Hytönen AM, Kaneko T, Lötvall J, Lindén A. 2003. A role of GM–CSF in the accumulation of neutrophils in the airways caused by IL-17 and TNFα. Eur Resp J. 21:387–393.
- Lambrecht BN, Hammad H. 2009. Biology of lung dendritic cells at the origin of asthma. Immunity. 31:412–424.
- Lang JE, Williams ES, Mizgerd JP, Shore SA. 2008. Effect of obesity on pulmonary inflammation induced by acute ozone exposure: role of IL-6. Am J Physiol. 294:L1013–L1020.
- Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, Blumenschein WM, Judo M, Ayanoglu G, McClanahan TK, et al. 2015. IL-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity. 43:727–738.
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔCt) method. Methods. 25:402–408.
- Lockhart E, Green AM, Flynn JL. 2006. IL-17 production is dominated by γδ T-cells rather than CD4 T-cells during Mycobacterium tuberculosis infection. J Immunol. 177: 4662–4669.
- Lukens JR, Barr MJ, Chaplin DD, Chi H, Kanneganti T-D. 2012. Inflammasome-derived IL-1β regulates production of GM-CSF by CD4+ T-cells and γδ T-cells. J Immunol. 188:3107–3115.
- Mathews JA, Williams AS, Brand JD, Wurmbrand AP, Chen L, Ninin FM, Si H, Kasahara DI, Shore SA. 2014. γδ T-cells are required for pulmonary IL-17A expression after ozone exposure in mice: Role of TNFα PLoS One. 9:e97707.
- Mathews JA, Wurmbrand AP, Ninin FM, Kasahara DI, Shore SA. 2013. IL-17A production by γδ T-cells is required for neutrophil and macrophage recruitment after subacute O3 exposure. Am Thor Soc Intl Conf. B95: A3510–A3510.
- Mathews P, Joel A, Wurmbrand AP, Ribeiro L, Neto FL, Shore S. 2014. Induction of IL-17A precedes development of airway hyper-responsiveness during diet induced obesity and correlates with complement factor D. Front Immunol 5:440.
- McKenna HJ, Stocking KL, Miller RE, Brasel K, De Smedt T, Maraskovsky E, Maliszewski CR, Lynch DH, Smith J, Pulendran B, et al. 2000. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. 95:3489–3497.
- Murakami M, Hibi M, Nakagawa N, Nakagawa T, Yasukawa K, Yamanishi K, Taga T, Kishimoto T. 1993. IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science. 260:1808–1810.
- Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, et al. 2002. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rβ1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 168:5699–5708.
- Roses RE, Xu S, Xu M, Koldovsky U, Koski G, Czerniecki BJ. 2008. Differential production of IL-23 and IL-12 by myeloid-derived dendritic cells in response to TLR agonists. J Immunol. 181:5120–5127.
- Shainheit MG, Smith PM, Bazzone LE, Wang AC, Rutitzky LI, Stadecker MJ. 2008. Dendritic cell IL-23 and IL-1 production in response to schistosome eggs induces TH17 cells in a mouse strain prone to severe immunopathology. J Immunol. 181:8559–8567.
- So A, de Smedt T, Revaz S, Tschopp J. 2007. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 9:R28.
- Stanek LW, Brown JS, Stanek J, Gift J, Costa DL. 2011. Air pollution toxicology – a brief review of the role of science in shaping the current understanding of air pollution health risks. Toxicol Sci. 120(S1):8–27.
- Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. 2009. IL-1 and IL-23 induce innate IL-17 production from γδ T-cells, amplifying TH17 responses and autoimmunity. Immunity. 31:331–341.
- Wands JM, Roark CL, Aydintug MK, Jin N, Hahn YS, Cook L, Yin X, Dal Porto J, Lahn M, Hyde D, et al. 2005. Distribution and leukocyte contacts of γδ T cells in the lung. J Leukocyte Biol. 78:1086–1096.
- Waskow C, Liu K, Darrasse-Jeze G, Guermonprez P, Ginhoux F, Merad M, Shengelia T, Yao K, Nussenzweig M. 2008. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol. 9:676–683.
- Zhang F, Huang G, Hu B, Qian GS, Song Y. 2015. Recombinant HMGB1 A box protein inhibits TH17 responses in mice with neutrophilic asthma by suppressing dendritic cell-mediated TH17 polarization. Int Immunopharmacol. 24:110–118.