Hereditary hemochromatosis (HH) is a not uncommon autosomal recessive and potentially life-threatening disease. The hemochromatosis gene was identified by Feder et al. in Citation1996. About 1 in 200 individuals is estimated to be homozygous for the most common mutation—C282Y/C282Y. In the classical form of the disease, cysteine is substituted by tyrosine at amino acid 282 in both alleles. The so-called compound heterozygoty is less common (representing about 10% of cases) but is also compatible with HH. Here, histidine is substituted by aspartic acid at amino acid 63 in one allele and cysteine by tyrosine at amino acid 282 in the other (C282Y/H63D).
Due to increased intestinal absorption, homozygotes develop iron overload but penetrance is very variable (McCune et al. Citation2006). Most orthopedic surgeons meet patients with undiagnosed HH on a yearly basis and an early diagnosis and treatment is important in order to avoid cirrhosis of the liver. Also, the risk of developing hepataocellular cancer is at least 20-fold higher than in their first-degree relatives (Elmberg et al. Citation2003). Fatigue and arthritis, unspecific and therefore often neglected, are common presentations whereas the classic bronze diabetes (darkened skin, diabetes, and cirrhosis) is the final stage of untreated HH that is very rarely seen (McDonell et al. Citation1999).
A survey of 2,851 patients with hemochromatosis showed that patients had consulted a physician after an average of 2 years of symptoms, and on average it took a further 10 years before the diagnosis was made (McDonell et al. Citation1999). This is unfortunate, since medical treatment—i.e. regular phlebotomy—is effective.
During the period 2001–2008, I replaced 93 ankles in 86 patients and 4 of these patients proved to have hemochromatosis. Here I have reviewed the orthopedic manifestations in 7 patients who had one or both ankles replaced during this period, 3 of whom had had their ankles replaced elsewhere.
Cases
The original records from all orthopedics departments and units of gastroenterology involved were consulted and the author interviewed the patients at least twice.
All but 1 patient were below the age of 45 when they had their first joint symptoms, and there was usually a long patient’s and doctor’s delay before diagnosis and treatment. In 4 cases the hip, knee or ankle joint, and not the MP joints, was the first joint to become symptomatic. Only in one case (patient 5) did the joint symptoms start after the time that the diagnosis of HH had been made ().
Table 1. Demographic data, laboratory findings, and joints replaced in 7 patients with hereditary hemochromatosis
4 patients (cases 1–4) had been referred to me from other hospitals due to painful osteoarthrosis of one or both ankles. In 3 of these cases, the diagnosis had been made before referral by laboratory testing and liver biopsy but the genotype was determined at referral.
In patient 3, I suspected and confirmed the diagnosis. She proved to have a compound heterozygoty (C282Y/H63D). The patient had donated blood regularly, which explains her only modestly increased laboratory values. 3 other patients with HH (cases 5–7) who had been operated on for a total ankle prosthesis elsewhere were identified via the Swedish National Board of Health and Welfare. The correct diagnosis had been reported to the Swedish Ankle Arthroplasty Register (Henricson et al. Citation2007) in only one of these 3 cases.
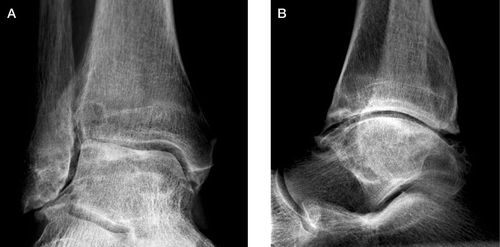
None of the patients had reported any major ankle trauma. Preoperative radiographs of all ankles that had been replaced, and the as yet unreplaced ankles in cases 5 and 6, were scrutinized. The radiographic pattern was uniform, with reduction of the joint space, bony eburnation, cysts in the distal tibia and/or talus, and osteophytes. The latter were always located anteriorly at the neck of the talus and distal tibia, but sometimes also posteriorly. With few exceptions, the reduction of the joint space was located laterally and always anteriorly.
Case no 2. The right ankle preoperatively. This man had his right ankle and left knee replaced during the same anesthesia, at the age of 60. The following year his left ankle and right hip were replaced, also during the same anaesthesia. Pain and swelling of the knee started at the age of 40, but 13 years later it was first confirmed that he suffered from hemochromatosis. The ankles became symptomatic at the age of 42. Reduction of the joint space is seen anteriorly and laterally and there are osteophytes, notably anteriorly at the neck of the talus and distal tibia. Bony eburnation and cysts in the talus and distal tibia can also be seen.
Discussion
Joint complaints are one of the most frequent symptoms of HH and they are often the first clinical manifestation of disease (McDonell et al. Citation1999, Inês et al. Citation2001, von Kempis Citation2001, Rihl and Kellner Citation2004). Although cirrhosis of the liver and cancer are important for mortality in patients with HH, arthropathy has the greatest effect on quality of life (von Kempis Citation2001).
The mechanism behind the arthropathy in HH is unknown. In HH, the sensitivity to cartilage damage is presumably increased and/or reparative capacity is reduced (Huch et al Citation1997). In most studies, no correlation has been found between serum ferritin levels and arthropathy. In contrast to other manifestations of the disease, the joint symptoms do not diminish after phlebotomy, or if so, only to a minor degree (Adams et al Citation1997, Sinigaglia et al Citation1997, McDonell et al. Citation1999, Rihl and Kellner Citation2004). Valenti et al. (Citation2008) have, however, reported that 32 of 88 patients with phenotypically expressed HH had radiographically verified changes in the metacarpophalangeal (MCP) joints, and that the severity of these changes was influenced by the degree of iron overload.
In textbooks and most papers on HH, symptoms from—and radiographic changes in—the MCP joints are repeatedly described as typical for the disease, whereas little or no attention has been drawn to other joints. The reason for this is presumably that degenerative changes of other joints are much more common and cannot be distinguished from those that occur in HH.
Whether or not major joints (i.e. the hip, knee, ankle, shoulder, and elbow) are affected more often in HH than in the general population is unclear. Lunn et al. (Citation2005) found 2 out of 116 patients who had undergone primary hip replacement to be C282Y homozygotes—a figure that is similar to the prevalence in the general population of Ireland. Curiously enough, substantially more patients—10 of 101 patients—who underwent revision of their hip were C282Y homozygotes. In a large population study, Adams et al. (Citation2005) only found self-reported arthritis to be more common in males who were homozygous for H63D than in participants without HFE mutations. Walker et al. (Citation2006) demonstrated an association between genotype and interleukin 1 receptor antagonist (IL1RN) levels in patients with HH and joint pain. Axford et al. (Citation1991) described an increased incidence of hip involvement, and Rollot et al. (Citation2005) suggested that osteonecrosis of the femoral head may be one expression of HH.
My observations and those of a few others (Bayley and Gardner 1998, Jacki et al. Citation1999, Schmid et al. Citation2003, Davies and Saxby Citation2006) indicate that the ankle may also be a key joint in HH. In the literature, I have found 5 papers that together reported on 13 patients who had had one ankle replaced and 3 papers that reported on HH patients who had had their hip joint replaced (). Here I report on another 7 patients who had 10 ankles and 6 other major joints replaced.
Table 2. HH and joint replacement as reported in the literature
For unknown reasons, the ankle is less susceptible to osteoarthritis than the knee and hip, and symptomatic osteoarthritis of the ankle is uncommon even at advanced age.
All the patients in my study had one or two ankles replaced, and all but one patient also had at least one other joint that was replaced or diseased. Recently, Carroll (Citation2006) reported a strong and statistically significant association between HFE gene mutations and primary OA in the ankle joint. Due to the frequent presence of OA of the second and third MCP joint in these patients, the same author also suggested the existence of a type-2 polyarticular OA phenotype that closely resembles the arthropathy of HH, which appears to be clinically differentiable from a type-1 OA or nodal generalized OA (NGOA).
There do not seem to be any radiographic phenomena that are typical of HH except for the reduction of joint space, bony eburnation, and broadening of the metacarpal heads seen in the MCP joints. This differs from what is usually seen in other types of arthritis.
The MRI appearance of arthropathy in HH cannot be distinguished from that of other types of degenerative osteoarthritis; the method cannot demonstrate the presence of iron in synovium, synovial fluid, or cartilage. It may be that the iron is below the threshold of detection by existing MRI techniques (Jager et al. Citation1997, Papakonstantinou et al. Citation2005). At surgery, the 7 ankles replaced by myself (cases 1–4) had the same appearance as in ordinary, degenerative osteoarthritis. In cases 2 and 4, the resected joint parts were examined histologically. No traces of iron were found.
The presence of long-standing joint pain and/or osteoarthritis in a person below the age of 55–60 years should thus arouse suspicion of HH if the symptoms cannot be related to another specific disease, e.g. seropositive rheumatoid arthritis, psoriasis, or arthritis urica. If more than one major joint is involved—notably bilateral ankle arthropathy without previous trauma—the suspicion is strengthened (Davies and Saxby Citation2006).
In such cases, plasma or serum iron levels, total iron-binding capacity (TIBC), and serum ferritin should be analyzed. An iron-saturation level (Fe/TIBC × 100) above 50% or an increased ferritin value should be followed by genetic testing. If this confirms that the patient is homozygous, or has a so-called compound heterozygoty for HH, the individual should be referred to a gastroenterologist for further examination. Joint symptoms do not appear to be influenced by early diagnosis and treatment, but awareness of the condition and positive screening may prevent patients and their relatives from undergoing the more serious consequences of HH by regular phlebotomy.
- Adams PC, Deugnier Y, Moirand R, Brissot P. The relationship between iron overload, clinical symptoms, and age in 410 patients with genetic hemochromatosis. Hepatology 1997; 25: 162–6
- Adams PC, Reboussin DM, Barton JC, McLaren CE, Eckfeldt JH, McLaren GD, Dawkins FW, Acton RT, Harris EL, Gordeuk VR, Leiendecker-Foster C, Speechley M, Snively BM, Holup JL, Thomson E, Sholinsky P. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med 2005; 352: 1769–78
- Axford JS, Bomford A, Revell P, Watt I, Williams R, Hamilton EB. Hip arthropathy in genetic hemochromatosis. Radiographic and histologic features. Arthritis Rheum 1991; 34: 357–61
- Bailey EJ, Gardner AB. Hemochromatosis of the foot and ankle. Report of three cases and review of the literature. Clin Orthop 1998, 349: 108–15
- Carroll GJ. Primary osteoarthritis in the ankle joint is associated with finger metacarpophalangeal osteoarthritis and the H63D mutation in the HFE gene: evidence for a hemochromatosis-like polyarticular osteoarthritis phenotype. J Clin Rheumatol 2006; 12: 109–13
- Davies MB, Saxby T. Ankle arthropathy of haemochromatosis: a case series and review of the literature. Foot Ankle Int 2006; 27: 902–6
- Elmberg M, Hultcrantz R, Ekbom A, Brandt L, Olsson S, Olsson R, Lindgren S, Lööf L, StÅl P, Wallerstedt S, Almer S, Sandberg-Gertzén H, Askling J. Cancer risk in patients with hereditary hemochromatosis and in their first-degree relatives. Gastroenterology 2003; 125: 1733–41
- Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Gen 1996; 13: 339–408
- Fevang B-T, Lie S, Havelin L, Brun J, Skredderstuen A, Furnes O. 257 ankle arthroplasties performed in Norway between 1994 and 2005. Acta Orthop 2007; 78: 575–83
- Henricson A, Skoog S, Carlsson Å. The Swedish ankle arthroplasty register. An analysis of 531 arthroplasties between 1993 and 2005. Acta Orthop 2007; 78: 569–74
- Hinterman B. Short- and mid-term results with the STAR total ankle prosthesis. Orthopäde 1999; 28: 792–803, (In German)
- Hosman AH, Mason RB, Hobbs T, Rothwell AG. A New Zealand national joint registry review of 202 total ankle replacements followed for up to 6 years. Acta Orthop 2007; 78: 584–91
- Huch K, Kuettner KE, Dieppe P. Osteoarthritis on the ankle and knee joints. Semin Arthritis Rheum 1997; 26: 667–74
- Inês LS, da Silva JA, Malcata AB, Porto AL. Arthropathy of genetic hemochromatosis: a major and distinctive manifestation of the disease. Clin Exp Rheumatol 2001; 19: 98–101
- Jacki SH, Uhl M, Adler CP, Peter HH, von Kempis J. Predominant ankle arthropathy in hereditary haemochromatosis. Rheumatology Oxford 1999; 38: 378–9
- Jager HJ, Mehring U-M, Gotz GF, Neise M, Erlemann R, Kapp HJ, Mathias KD. Radiological features of the visceral and skeletal involvement of hemochromatosis. Eur Radiol 1997; 7: 1199–206
- Jarde O, Gabrion A, Meire P, Trinquir-Lautard JL, Vives P. Complication and failures of total ankle prosthesis. Apropos of 21 cases. Rev Chir Orthop Reparatrice Appar Mot 1997; 83: 645–51, (In French)
- Lecoules S, el Maghraoui A, Damiano J, Lechevalier D, Magnin J, Eulry F. Hip arthroplasty in genetic hemochromatosis. Report of 5 cases. Rev Med Interne 2002; 23: 454–9, (In French)
- Lunn JV, Gallagher PM, Hegarty S, Kaliszer M, Crowe J, Murray P, Bouchier-Hayes D. The role of hereditary hemochromatosis in aseptic loosening following primary total hip arthroplasty. Orthop Res 2005; 23: 542–8
- McCune CA, Ravine D, Carter K, Jackson HA, Hutton D, Hedderich J, Krawczak M, Worwood M. Iron loading and morbidity among relatives of HFE C282Y homozygotes identified either by population genetic testing or presenting as patients. Gut 2006; 55: 554–62
- McDonell S, Preston B, Jewell S, Barton J, Edwards C, Adams PC, Yip R. A survey of 2,851 patients with haemochromatosis: Symptoms and response to treatment. Am J Med 1999; 106: 619–25
- Montgomery KD, Williams JR, Sculco TP, DiCarlo E. Clinical and pathologic findings in hemochromatosis hip arthropathy. Clin Orthop 1998, 347: 179–87
- Papakonstantinou O, Mohana-Borges AVR, Campell L, Haghighi P, Resnick D. Hip arthropathy in a patient with primary hemochromatosis: MR imaging findings with pathological correlation. Skeletal Radiology 2005; 34: 180–4
- Rihl M, Kellner K. Die Artopathie der Heriditären Hämochromatose. Z Rheumatol 2004; 63: 22–9
- Rollot F, Wechsler B, du Boutin le TH, Degennes C, Amoura Z, Hachulla E, Piette JC. Hemochromatosis and femoral head aseptic osteonecrosis: a nonfortuitous association?. J Rheumatol 2005; 32: 376–8
- Schmid H, Struppler C, Braun GS, Kellner W, Kellner H. Ankle and hindfoot arthropathy in hereditary hemochromatosis. J Rheumatol 2003; 30: 96–9
- Sinigaglia L, Fargion S, Fracanzani AL, Binelli L, Battafarano N, Varenna M, Piperno A, Fiorelli G. Bone and joint involvement in genetic hemochromatosis: role of cirrhosis and iron overload. J Rheumatol 1997; 24: 1809–13
- Valenti L, Rametta R, Dongiovanni P, Maggioni M, Ludovica Fracanzani A, Zappa M, Lattuada E, Roviaro G, Fargion S. The hand arthropathy of hereditary hemochromatosis is strongly associated with iron overload. J Rheumatol 2008; 35: 153–8
- von Kempis J. Arthropathy in hereditary hemochromatosis. Curr Opin Rheumatol 2001; 13: 80–3
- Walker EJ, Riddell J, Rodgers HJ, Bassett ML, Wilson SR, Cavanaugh JA. IL1RN genotype as a risk factor for joint pain in hereditary haemochromatosis. Ann Rheum Dis 2006; 65: 271–2