Abstract
The 46,XX testicular disorder of sex development (46,XX testicular DSD) is a rare phenotype associated with disorder of the sex chromosomes. We describe the clinical, molecular, and cytogenetic findings of a 16- and a 30-year-old male patient with sex-determining region Y (SRY)-positive 46,XX testicular DSD. Chromosomal analysis revealed 46,XX karyotype. Fluorescence in situ hybridization (FISH) showed the SRY region translocated to the short arm of the X chromosome. The presence of the SRY gene was also confirmed by polymerase chain reaction (PCR). The X chromosome inactivation (XCI) assay showed that both patients have a random pattern of X chromosome inactivation. This report compares the symptoms and features of the SRY-positive 46,XX testicular DSD patients.
Introduction
46,XX testicular disorder of sex development [46,XX testicular DSD; 46,XX gonadal dysgenesis; XX male syndrome (MIM 400045)] was described by De la Chapelle et al. in 1964 [De La Chapelle et al. Citation1964]. It is a rare disorder with an incidence of 1/20,000 males [De La Chapelle Citation1981]. Approximately 80% of the cases are sex-determining region Y (SRY)-positive, while the remaining cases are SRY-negative [Vilain Citation2003]. The presentation includes primary hypogonadism, small testes, gynecomastia, and hyalinization of seminiferous tubules, azoospermia, and infertility [Ergun-Longmire et al. 2005]. In this study, we report the clinical, cytogenetical, fluorescence in situ hybridization (FISH), and molecular findings of two cases with SRY-positive 46,XX testicular DSD.
Clinical Report
Patient I
A 30-year-old male was referred from Urology Clinic with a five year history of male infertility. His wife had a healthy child from her first marriage. He has two healthy fertile brothers and one healthy sister. He presented a normal male phenotype with short stature, relatively poor beard, pubic and axillary hair development, gynecomastia, stretched penile length of 14 cm, small testes and normal intelligence ().
Table 1. Clinical findings of patients.
He was treated with human chorionic gonadotropin for six months and with clomiphene citrate in another clinic nine years ago. He had bilateral mastectomy eight years ago. Abdominal pelvic ultrasonography (USG) showed atrophic seminal vesicles and prostate and no mullerian derivatives.
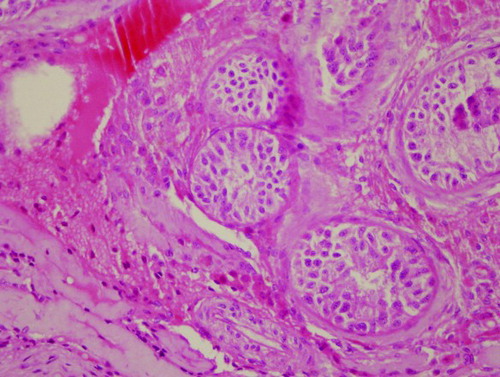
Microscopic testicular sperm extraction (TESE) was performed. Testicular biopsies were taken from four different parts of the testes. After mechanical separation of seminiferous tubules, microscopic examination revealed no live or dead spermatozoa. Histopathological examination revealed an apparent thickening of the basal membrane and only Sertoli cells. There were no spermatogenetic cells inside the lumen of seminiferous tubules. Extensively hyalinised seminiferous tubules were present, consistent with a Sertoli-cell-only syndrome ().
Figure 1. Histopathological results of testis biopsies of patient 1. Testicular histology shows an apparent decrease of seminiferous tubules in testicular parenchyma. Most of the seminiferous tubules show seminiferous tubule hyalinization and Sertoli-cells only syndrome with complete absence of spermatogenetic cells (Haemotoxilin-eosin x400 magnification).
Patient II
Patient II was a 16-year-old male admitted to the Urology Clinic with complaints of having small testes and penis. He was the second child of healthy parents. He has a 19 year old healthy brother. Past medical history was unremarkable with respect to probable risk factors such as trauma, medication, radiation, infection, or chemical exposure. He was born after a normal pregnancy period and delivery. His birth weight was 3,700 g and he reached puberty at the age of 15. At admission, his height was 152 cm (-3 SD). He had a normal scrotum and a stretched penile length of 12.5 cm (Marshall & Tanner stage 2) [Marshall and Tanner Citation1970]. Testicular volume was 6 ml on the left and right and a vas deferens was palpable bilaterally. Testosterone replacement treatment with depot testosterone undecanoate was started due to hypogonadism. Table I presents the clinical findings of both patients.
Results and Discussion
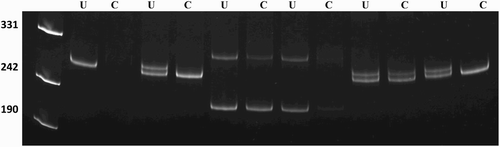
Chromosomal analysis using GTG-banding revealed a 46,XX karyotype (450 bands of resolution) and both patients were diagnosed as XX males. PCR analysis showed amplification of sY1532, SRY, and ZFY genes that are located on the short arm of the Y chromosome (, ). None of the tested AZFa, AZFb, and AZFc sequences could be detected. FISH analysis confirmed the presence of SRY translocated to the Xp region demonstrating a karyotype 46,XX. ish(DXZ1x1)[6]/(DXZ1x2,SRYx1)[243]/(DXZ1x3,SRYx2)[4] for patient I, and 46,XX.ish der(X)t(X;Y)(p22.3;p11.3)(SRY+) for patient II (, ). Methylation analysis of the AR gene of both cases demonstrated random X chromosome inactivation (XCI) (53%:47% and 59%:41% skewing, respectively) in peripheral blood sample and 59% in testicular tissue in patient I ().
Figure 2. Electrophoresis of the PCR products of SRY+ chromosomes. Line 1, positive control (male); line 2, marker (R322DNA/BsuRI); line 3, SRY and ZFX/Y amplification of case I; line 4, SRY and ZFX/Y amplification of case II; line 5, female control; line 6, negative control.
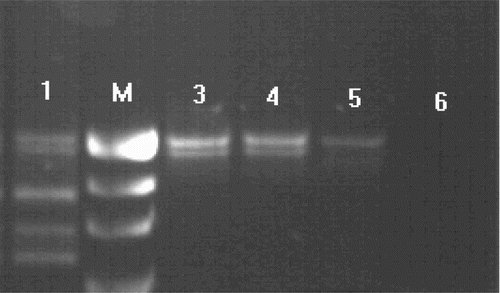
Table 2. Chromosomal and molecular analysis.
In this study, we describe the clinical, cytogenetic, and molecular features of two males with SRY-positive 46,XX testicular DSD. Both cases have the appearance of classical 46,XX, SRY positive male phenotypic features, namely, azoospermia, decreased body height, and small testis. In comparison, some of 46,XX SRY-positive patients may present cryptorchidism or can have genital ambiguities and can even be true hermaphrodites. The variation of the phenotypic features of the SRY positive 46,XX males might be related with the length of the Y chromosomal material translocated on the short arm of the X chromosome [Bouayed Abdelmoula et al. Citation2003; Kusz et al. Citation1999], X inactivation pattern [Bouayed Abdelmoula et al. Citation2003; Kusz et al. Citation1999; Boucekkine et al. Citation1985; Schempp et al. Citation1989], position effect [Sharp et al. Citation2005] or presence of another Y linked gene [Bouayed Abdelmoula et al. Citation2003] which may have a role in male sexual development at the translocated segment.
Obligatory crossing over may take place between the X and Y chromosomes at the pseudoautosomal regions (PARs) during male meiosis. Along the non-recombining region of the Y chromosome, there are regions that have homology with the X chromosome but these regions do not normally undergo recombination [McElreavey and Cortes Citation2001]. The SRY gene encodes the ‘testis-determining factor’ which is essential for the development of the male during fetal life [Sinclair et al. Citation1990]. In some cases, a Y chromosome fragment can be translocated to the X chromosome by an aberrant interchange between the X and the Y chromosomes during paternal meiotic division [Ferguson-Smith Citation1966; Margarit et al. Citation2000]. The reason for this translocation could be the homology existing between the two homologous genes, PRKX and PRKY. The sequence similarity between these genes is 94%. The PRKY gene is approximately 7 Mb away from the obligatory recombination region, PAR1 [Weil et al. Citation1994; Schiebel et al. Citation1997]. As a result of that recombination, the individuals have male sexual characteristics because of the presence of SRY gene on the X chromosome despite the XX sex chromosome pattern. The size of the translocated fragment is variable between SRY-positive XX males [Sharp et al. Citation2005; Vergnaud et al. Citation1986]. We did not determine in any of our cases the length of the translocated fragment.
The presence of the SRY gene on Xp leads to normal male sexual development. Though 46,XX testicular DSD patients usually have normal external genitalia, sometimes hypospadia or cryptorchidism may be seen [Yencilek and Baykal 2005]. Therefore, it is difficult to find 46,XX testicular DSD males before puberty. Our cases had no sexual ambiguities and were diagnosed after puberty [Wang et al. 2009]. Occasionally, SRY-positive XX males have sexual ambiguity despite having the SRY gene depending on the amount of material of Yp transferred to the X chromosome [Kusz et al. Citation1999; Queipo et al. Citation2002].
In 46,XX testicular DSD patients, the level of testosterone is normal during puberty but deficiency develops during adulthood leading to hypergonadotropic hypogonadism. In addition, infertility, and high levels of LH and FSH are observed [Yencilek and Baykal 2005]. In patient II, the testosterone level was mildly suppressed and a further decrease in the testosterone level is expected as presented in patient I. Microscopic examination of both testes showed hyalinization of most tubules in patient I without any spermatogenetic cells, consistent with, Sertoli-cell-only syndrome [Bouayed Abdelmoula et al. Citation2003].
The XCI assay was used to determine whether preferential X-chromosome inactivation could be detected. Both patients showed a random inactivation pattern. The results of the XCI analysis in 46,XX testicular DSD patients are controversial [Bouayed Abdelmoula et al. Citation2003; Kusz et al. Citation1999; Boucekkine et al. Citation1985; Schempp et al. Citation1989; Vorona et al. Citation2007]. Both random and nonrandom X chromosome inactivation patterns have been reported. It has been suggested that there is no apparent relationship between phenotype and the pattern of X inactivation in SRY-positive 46,XX male patients. Additionally, the studied tissue can not fully represent all the tissues. According to our knowledge in most cases XCI analysis was performed on peripheral blood. XCI analysis was performed for both peripheral blood and testes for patient I to study tissue-specific patterns of X inactivation. Our results revealed that there is no preferential silencing of der(X;Y) in peripheral blood and testes. Differences in the size of the translocated fragment could lead to discrepancies of X chromosome inactivation analysis [Allen et al. Citation1992; Schempp et al. Citation1989; Queipo et al. Citation2002]. The length of the Y chromosome fragment in 46,XX male patients ranges from 40 kb to > 75% of the Yp. The phenotypic differences could depend on the proximity of the breakpoint to the SRY gene. The presence or absence of cryptic recombinations may affect the expression of SRY gene by position effect [Sharp et al. Citation2005].
Recently Chernykh and colleagues [2009] reported X chromosomal mosaicism in a 37-year-old SRY positive 46,XX male. Low grade mosaicism with three different cell lines was detected in patient I. It has been suggested that the presence of structurally normal and abnormal X chromosomes predisposes one to mitotic nondisjunction that can lead to X chromosomal mosaicism [Chernykh et al. Citation2009]. Abnormal mitotic segregation would be related with advanced age and cause X chromosomal mosaicism [Ogata et al. Citation2001] as it was in patient I.
Figure 3. Fluorescence in situ hybridization (FISH) results. FISH analysis was performed on both interphase nuclei and metaphase spreads of patient I and on metaphase spreads of patient II. A-B) Interphase cells: A) 3 copies of DXZ1 and 1 copy of sex-determining region Y (SRY) (on top) and 2 copies of DXZ1 and 1 copy of SRY (on bottom), B) 2 copies of DXZ1 and 1 copy of SRY (on top) and only 1 copies of DXZ1 (on bottom); C-E) Metaphase cells: C) 2 copies of DXZ1 and 1 copy of SRY, D) 3 copies of DXZ1 and 1 copy of SRY, and E) 2 copies of DXZ1 and 1 copy of SRY (patient II). DXZ1:
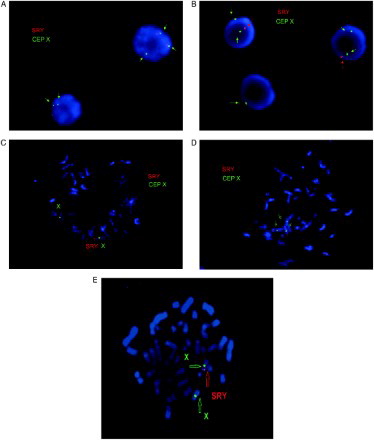
Figure 4. X chromosome inactivation pattern of case I and case II. PCR products of undigested (U) and HpaII-digested (C) DNA from peripheral blood and testis (Case 1). XCI patterns of the cases. Line 1, marker (pUC mix 8), 331, 242 and 190 bp fragments are visible; line 2 and 3: male control; line 4 and 5, case I (PB); line 6 and 7, case I (T); 8 and 9 case II (PB); line 10 and 11, positive control. PB: Peripheral blood, T: Testis.
In summary, we report two XX males with SRY gene at the end of the short arm of chromosome X. Both presented a classical phenotype and a normal XCI suggesting that skewed X inactivation is not mandatory for the distinct clinical signs of the disease.
Materials and Methods
Prior to the study, all patients were informed about the investigation and a signed consent form was obtained. All protocols involving the use of human tissue specimens were approved by the Institutional Review Board.
Cytogenetic analysis
Chromosomal analyses of peripheral blood samples were performed using standard procedures. Chromosome banding was achieved by Giemsa (GTG) method described in detail [Seabright Citation1971].
FISH analysis
FISH analysis was performed on both interphase nuclei and metaphase spreads of patient I and only on metaphase spreads of patient II (the supplement section contains details on FISH analysis of a third patient). FISH was performed on metaphase spreads and interphase nuclei of the lymphocytes, according to the manufacturer's instructions. X centromere (CEP X) and SRY-specific (LSI SRY) probes, (Vysis Inc., Downers Grove, IL, USA) were used.
Molecular analysis
The genomic DNA was isolated from peripheral venous blood samples of the patients by the ‘salting out’ method [Miller et al. Citation1988] and from testes using a commercially available kit (Invitrogen Genomic DNA Mini Kit, Carlsbad, CA, USA) according to the manufacturer's instructions. In addition to a negative control, normal male DNA and normal female DNA were used as controls. SRY (sY14 and sY1532), ZFY, sY84, sY86, sY127, sY134, sY254, sY255, SMCY, and CDY2 locus amplification was performed using multiplex PCR (Simoni et al. Citation2004; Nakashima et al. Citation2002). The amplification products were resolved on 2% agarose and analyzed using a video gel documentation system (Biolabs, Kyoto, Japan) after staining with ethidium bromide.
X-chromosome inactivation analysis
DNA was isolated from blood sample of the patient using Nucleospin Blood kit (Macharey-Nagel, Düren, Germany) as in the protocol supplied by the manufacturer. X chromosome inactivation (XCI) pattern of the patient is determined by genotyping of the highly polymorphic CAG repeat in the androgen receptor (AR) gene, known as XCI assay, as in the protocol [Ozbalkan et al. Citation2005; Allen et al. Citation1992]. Briefly, DNA was digested with methylation specific restriction enzyme HpaII (MBI Fermentas, Vilnius, Lithuania), which cleaves unmethylated (or active) X fragments. Therefore, the methylated (or inactive) X chromosome remains intact for PCR amplification. PCR products were separated on 8% denaturing 29:1 acrylamide:bisacrylamide gel for 4 h at constant 15W. MultiAnalyst version 1.1 software (Bio-Rad, Hercules, California) was used for densitometric analysis of the alleles.
Testicular histology
Testicular biopsy was taken during microscopic TESE under spinal anesthesia in the operating room. The specimen fixed in Bouin's solution was delivered to the pathology laboratory. Following the fixation, routine procedure was performed and sections were stained with Haemotoxilin-eosin (H&E) [Magid et al. Citation1990].
Declaration of interest: The authors report no conflict of interest.
Author contributions: Preparation of the manuscript, molecular and cytogenetic analysis: SG; Preparation of the manuscript, clinical evaluation of the patients: RA; Director of Cytogenetics Laboratory: GO; Preparation of testis biopsy and histological evaluation: OA; Preparation of some parts of the manuscript, clinical evaluation of the patients: FA; XCA and preparation of related parts in manuscript: OEO; FISH analysis and editing of manuscript: GO; XCA and editing of manuscript: TO; Editing of manuscript: HB.
Note of added Proof
The X chromosome inactivation (XCI) assay of a third patient showed a non-random pattern of X chromosome inactivation (Supplemental – 4 and Supplemental Tables I and II). As detailed in the Supplement we report three XX males with SRY gene at the end of the short arm of chromosome X. All patients presented a classical phenotype with normal and completely skewed XCI suggesting that skewed X inactivation is not mandatory for the distinct clinical signs of the disease.
SUPPLEMENT
Two Males with SRY-Positive 46,XX Testicular Disorder of Sex Development
S. Gunes, R. Asci, G. Okten, F. Atac, O. E. Onat, G. Ogur, O. Aydin, T. Ozcelik, and H. Bagci
A third recent case describing the clinical, molecular, and cytogenetical findings of a 22-year-old male patient with SRY-Positive 46,XX testicular DSD is presented. Chromosomal analysis revealed 46,XX karyotype. The X chromosome inactivation (XCI) assay showed a non-random pattern of X chromosome inactivation. The 22-year-old male was referred to the Urology Clinic with 1-year history of intermittent scrotal pain. He has one healthy brother and a sister. Past medical history was unremarkable. He was born after a normal pregnancy period and delivery. His birth weight was 3,800 g and he reached puberty at the age of 15. He presented a normal male phenotype with normal stature and height, relatively poor beard, normal pubic and axillary hair development, gynecomastia, small testis, and normal intelligence (Supplemental ). His height was 172 cm. He had a normal scrotum and a stretched penile length of 13 cm (Marshall & Tanner stage 4). Testicular volume was 5 mL on the left and right and bilateral vas deferens was palpable. The testosterone level was mildly suppressed.
Supplemental Table I. Clinical findings of Patient III
Chromosomal analysis by using GTG-banding revealed a 46,X,der(X)t(X;Y)(p22.3;p11.3) for patient III (550 band resolution) (Supplemental Fig. 1) diagnosed an XX males. PCR analysis showed amplification of sY1532, SRY and ZFY genes that are located on the short arm of the Y chromosome (Supplemental Fig. 2, Supplemental ). None of the tested AZFa, AZFb, and AZFc sequences could be detected.
Supplemental Table II Chromosomal and molecular analysis.
FISH analysis confirmed the presence of SRY locus translocated to the Xp region demonstrating a karyotype 46,XX.ish der(X)t(X;Y)(p22.3;p11.3)(SRY+) for patient III (Supplemental , Supplemental Fig. 3). Patient III exhibited completely skewed XCI pattern with a skewed X chromosome inactivation ratio of 100% (Supplemental Fig. 4).
The height of the patient III is above those of the two patients previously reported. Besides a completely skewed XCI pattern of patient III, the karyotype of patient III demonstrated translocated fragment of Y chromosome. The phenotypic differences could depend on the proximity of the breakpoint to the SRY gene. The presence or absence of cryptic recombinations may affect the expression of SRY gene by position effect [Sharp et al. 2005]. Alternatively, Vorona and colleagues [2007] reported SRY translocations may lead to disturbances in the expression of the short stature homebox (SHOX) gene which is located in the pseudoautosomal region of the X chromosome. All patients presented a classical phenotype with normal and completely skewed XCI suggesting that skewed X inactivation is not mandatory for the distinct clinical signs of the disease.
Abbreviations
| 46,XX testicular DSD: | = | 46,XX testicular disorder of sex development |
| SRY: | = | sex-determining region Y |
| FISH: | = | fluorescence in situ hybridization |
| PCR: | = | polymerase chain reaction |
| XCI: | = | X chromosome inactivation |
| PARs: | = | pseudoautosomal regions |
| DXZ1: | = | X centromere a-satellite probe. |
Supplementary Material
Download PDF (139.1 KB)References
- Allen, R.C., Zoghbi, H.Y., Moseley, A.B., Rosenblatt, H.M. and Belmont, J.W. (1992) Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 51:1229-1239.
- Bouayed Abdelmoula, N., Portnoi, M.F., Keskes, L., Recan, D., Bahloul, A., Boudawara, T., (2003) Skewed X-chromosome inactivation pattern in SRY positive XX maleness: a case report and review of literature. Ann Genet 46:11-18.
- Boucekkine, C., Nafa, D., Casanova-Bettane, M., Latron, F., Fellous, M. and Benmiloud, M. (1985) Evidence of a preferential inactivation of the paternally derived X chromosome in a 46,XX true hermaphrodite. Hum Genet 69:91-93.
- Chernykh, V.B., Kurilo, L.F., Shilova, N.V., Zolotukhina, T.V., Ryzhkova, O.P., Bliznetz, E.A., (2009) Hidden X chromosomal mosaicism in a 46,XX male. Sex Dev 3(4):p183-187.
- De La Chapelle, A., Hortling, H., Niemi, M. and Wennström J (1964) XX sex chromosomes in a human male. First case. Acta Med Scand 412:25-38.
- De La Chapelle, A. (1981) The etiology of maleness in XX men. Hum Genet 58:105–116.
- Ergun-Longmire, B., Vinci, G., Alonso, L., Matthew, S., Tansil, S., Lin-Su, K., (2005) Clinical, hormonal and cytogenetic evaluation of 46,XX males and review of the literature. J Pediatr Endocrinol Metab 18:739-748.
- Ferguson-Smith, M.A. (1966) X-Y chromosomal interchange in the aetiology of true hermaphroditism and of XX Klinefelter's syndrome. Lancet 288:475-476.
- Kusz, K., Kotecki, M., Wojda, A., Szarras-Czapnýk, M., Latos-Bielenska, A., Warenik-Szymankiewicz, A., (1999) Incomplete masculinisation of XX subjects carrying the SRY gene on an inactive X chromosome. J Med Genet 36:452-456.
- Magid, M.S., Cash, K.L. and Goldstein, M. (1990) The testicular biopsy in the evaluation of infertility. Semin Urol 8(1):p51-64.
- Margarit, E., Coll, M.D., Oliva, R., Gomez, D., Soler, A. and Ballesta, F. (2000) SRY gene transferred to the long arm of the X chromosome in a Y-positive XX true hermaphrodite. Am J Med Genet 90:25-28.
- Marshall, W.A. and Tanner, J.M. (1970) Variations in the pattern of pubertal changes in boys. Arch Dis Child 45:13-23.
- McElreavey, K. and Cortes, L.S. (2001) X-Y translocations and sex differentiation. Semin Reprod Med 19:133-139.
- Miller, S.A., Dykes, D.D. and Polesky, H.F. (1988) A simple salting out for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215.
- Nakashima, M., Koh, E., Namiki, M. and Yoshida, A. (2002) Multiplex sequence-tagged site PCR for efficient screening of microdeletions in Y chromosome in infertile males with azoospermia or severe oligozoospermia. Arch Androl 48(5):p351-351.
- Ogata, T., Matsuo, M., Muroya, K., Koyama, Y. and Fukutani, K. (2001) 47,XXX male: A clinical and molecular study. Am J Med Genet 98(4):p353-353.
- Ozbalkan, Z., Bagislar, S., Kiraz, S., Akyerli, B., Ozer, H.T., Yavuz, S., (2005) Skewed X chromosome inactivation in blood cells of women with scleroderma. Arthritis Rheum 52:1564-1570.
- Queipo, G., Zenteno, J.C., Peña, R., Nieto, K., Radillo, A., Dorantes, L.M., (2002) Molecular analysis in true hermaphroditism: demonstration of low-level hidden mosaicism for Y-derived sequences in 46,XX cases. Hum Genet 111:278-283.
- Schempp, W., Müller, G., Scherer, G., Bohlander, S.K., Rommerskirch, W., Fraccaro, M., (1989) Localization of Y chromosome sequences and X chromosomal replication studies in XX males. Hum Genet 81:144-148.
- Schiebel, K., Winkelmann, M., Mertz, A., Xu, X., Page, D.C., Weil, D., (1997) Abnormal XY interchange between a novel isolated protein kinase gene, PRKY, and its homologue, PRKX, accounts for one third of all (Y+) XX males and (Y-) XY females. Hum Mol Genet 6:1985-1989.
- Seabright, M. (1971) A rapid banding technique for human chromosomes. Lancet 2:971-972.
- Sharp, A., Kusz, K., Jaruzelska, J., Tapper, W., Szarras-Czapnik, M., Wolski, J., (2005) Variability of sexual phenotype in 46,XX(SRY+) patients: the influence of spreading X inactivation versus position effects. J Med Genet 42:420-427.
- Simoni, M., Bakker, E. and Krausz, C. (2004) EAA/EMQN best practice guidelines for molecular diagnosis of y-chromosomal microdeletions. State of the art 2004. Int J Androl 27(4):p240-240.
- Sinclair, A.H., Berta, P., Palmer, M.S., Hawkýns, J.R., Griffiths, B.L., Smith, M.J., (1990) A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 346:240-244.
- Vergnaud, G., Page, D.C., Simmler, M.C., Brown, L., Rouyer, F., Noel, B., (1986) A deletion map of the human Y chromosome based on DNA hybridization. Am J Hum Genet 38:109-124.
- Vilain, E.J. (2003) 46,XX Testicular Disorder of Sex Development. In: GeneReviews [Internet]. Pagon, R.A., Bird, T.C., Dolan, C.R., Stephens, K., ed. Seattle, (WA): University of Washington, Seattle.
- Vorona, E., Zitzmann, M., Gromoll, J., Schüring, A.N. and Nieschlag, E. (2007) Clinical, endocrinological, and epigenetic features of the 46,XX male syndrome, compared with 47,XXY Klinefelter patients. Clin Endocrinol Metab 92:3458-3465.
- Wang, T., Liu, J.H., Yang, J., Chen, J. and Ye, Z.Q. y(2009) 46, XX male sex reversal syndrome: a case report and review of the genetic basis. Andrologia 41:59-62.
- Weil, D., Wang, I., Dietrich, A., Poustka, A., Weissenbach, J. and Petit, C. (1994) Highly homologous loci on the X and Y chromosomes are hot-spots for ectopic recombinations leading to XX maleness. Nat Genet 7:414–419.
- Yencilek, F. and Baykal, C. y(2005) 46,XX male syndrome: a case report. Clin Exp Obst Gyn 32:263-264.