Abstract
We present a case of a 19-year-old phenotypically normal girl with premature ovarian failure. Cytogenetic analysis using G banding and fluorescence in situ hybridization (FISH) from cultured peripheral blood lymphocytes of the patient and the family revealed a de novo X;15 translocation and the imbalance to be 46,X,t(X;15)(Xpter → Xq21::15q11 → 15qter;15pter → 15q11::Xq21 → Xqter).
ish (CEPX+, wep15+, ISNRPN+, PML+, D15S10+, wcp15-, SNRRN-, PML-)[20]. The X chromosome inactivation (XCI) assay revealed a completely skewed XCI pattern in which selective pressure favors an active maternal allele. The Affymetrix 2.7 M cytogenetics whole-Genome array confirmed the chromosomal imbalance and identified disruption of the HDX gene at Xq21, the translocation breakpoint.
Introduction
Premature ovarian failure (POF; POF1: MIM 311360, POF2: MIM 300511; POF3: MIM 608996) [Beck-Peccoz and Perzani 2006], premature ovarian insufficiency (POI), are characterized by absences of menstruation (primary amenorrhea) or premature depletion of ovarian follicles with elevated gonadotrophin and low estrogen before the age of 40 years (secondary amenorrhea) [Cordts et al. Citation2011]. Iatrogenic intervention, autoimmune diseases, infections, X chromosome defects, and monogenic defects are possible causes of POF [Beck-Peccoz and Perzani 2006]. There are different types of X chromosome aberrations. These include monosomy X, trisomy X, partial deletions, and X;autosome translocations. X;autosome translocations are rare chromosomal abnormalities with an incidence rate of 1 in 30,000 in live births [Speroff and Fritz Citation2005].
Premature ovarian failure is clearly a heterogeneous disorder associated with mutations in FMR1 [POF1, (OMIM: 311360)] on Xq27.3, DIAPH2 [POF2A, OMIM: 300511) on Xq21.33 and POF1B [POF2B, (OMIM: 300604) on Xq21.1, FOXL2 [POF3, (608996)] on 3q22.1, BMP15 [POF4, (OMIM:300510)] on Xp11.22, NOBOX [POF5, (OMIM:611548)] on 7q35, NIGLA [POF6, (OMIM:612310)] on 2p13.3, and NR5A1 [POF7, (OMIM:612964)] on 9q33.3. In this paper, we report a phenotypically normal girl with an X;15 translocation that interrupts the highly divergent homeobox (HDX) (RefSeq accession number: NG_017158.1) gene on Xq21.1.
Case Report
A 17-year-old girl with secondary amenorrhea was admitted to the Department of Medical Biology, Section of Medical Genetics at Ondokuz Mayis University. She was the fourth child of healthy non-consanguineous parents. Her body height was 158 cm, and her weight was 51 kg. She menstruated at age of 12 and then at 17 years of age with continued hormone therapy. Ultrasonography (USG) revealed small ovaries with arrested folliculogenisis. Laboratory investigations demonstrated that her hormonal levels were as follows: follicle-stimulating hormone (FSH), 75.6 mU/mL; luteinizing hormone (LH), 100.5 mIU/mL; prolactin, 16.94 ng/mL, and estradiol, 83 pg/mL. FSH and LH were higher than normal. The measurement of thyroid-stimulating hormone (TSH) levels was normal.
Results and Discussion
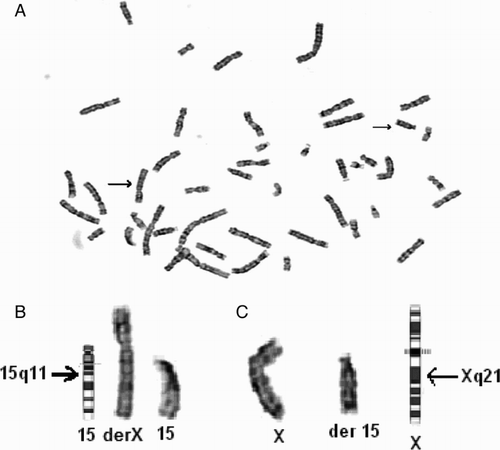
G banding cytogenetic analysis from cultured peripheral blood lymphocytes of the proband revealed a karyotype: 46,X,t(X;15)(q21;q11) ( A,B,C). The karyotypes of her mother, father, brother, and sisters were normal. FISH analysis has verified the de novo chromosomal imbalance to be 46,X,t(X;15)(Xpter→Xq21::15q11→15qter;15pter→ 15q11::Xq21→Xqter)
Figure 1. Cytogenetic analysis using G-banding assay from cultured peripheral blood lymphocytes. A) Metaphase image of the patient; upper arrow: der(15) chromosome and lower arrow: der(X) chromosome; B) Chromosome 15 ideogram, der(X)(Xpter → Xq21::15q11 → 15qter) chromosome, chromosome 15, and C) Chromosome 15, der (15)(15pter → 15q11::Xq21 → Xqter) chromosome, and X chromosome ideogram.
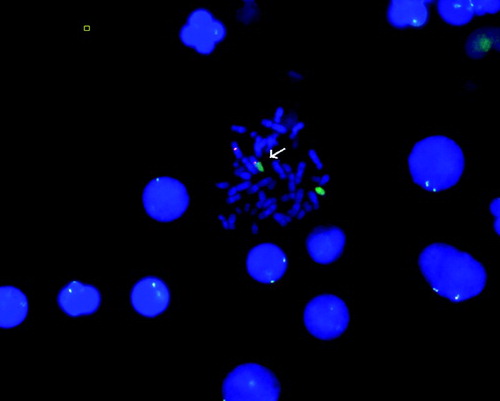
ish(CEPX+,wep15+, ISNRPN+, PML+, D15S10+, wcp15-, SNRRN-, PML-)[20] ().
Figure 2. FISH analysis of the patient showing t(X;15). FISH analysis was performed on metaphase spreads of the patient. Red signals show chromosome X centromere and green signals show chromosome 15. Normal X chromosome (red) and chromosome 15 (green) are evident. The arrow indicates derivative (X) chromosome.
Following conventional G-banding and FISH analysis, genome wide cytogenetic array analysis (Affymetrix Cytogenetics 2.7M Whole-Genome array) was used to determine the precise location and size of genomic imbalance. Cytogenetic array results showed the disruption of HDX gene at the translocation breakpoint region on the X chromosome (). XCI assay in the family revealed a completely skewed XCI pattern in the patient and random XCI patterns in the sisters as expected (). She was homozygous for the Familial Mediterranean fewer E148Q mutation.
Figure 3. Representation of the translocation breakpoint on X chromosome using Affymetrix
Cytogenetics Whole-Genome 2.7M Array. Analysis was performed by Affymetrix GeneChip.
Cytogenetics Whole-Genome 2.7M Array using default settings for image analysis, filtration, normalization, and annotation. Visualization of the graphical data was carried out using Chromosome Analysis Suite Software v1.0 software. At the translocated region on X chromosome highly divergent homeobox (HDX) gene is disrupted (represented by arrow).
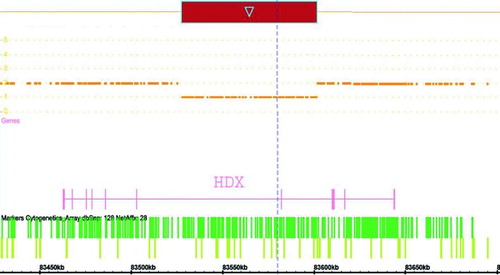
Figure 4. X-chromosome inactivation pattern of the family. Line 1, marker; line 2 and 3: brother of the patient; line 4 and 5, the patient; line 6,7,8,9, sisters of the patient; line 10 and 11 mother of the patient; line 12 and 13, father of the patient, and line 13 and 14, negative control. A single allele in the HpaII digested sample indicating non-random X-chromosome inactivation was observed in the patient (U: uncleaved and C: cleaved).

In the present study, we reported a patient with POF who has a de novo balanced t(X;15) translocation determined by G banding and FISH. Characterization of the transcription breakpoint using genome wide cytogenetics arrays revealed that the translocation interrupts the HDX (Refseq accession number: NG_017158.1) gene on Xq21. HDX is a known protein coding gene with 3 isoforms that encodes a highly divergent homeobox protein with unknown function and is not associated with any disease.
Cytogenetic studies have suggested that the long arm of the X chromosome is mainly involved in defects of ovulation [Bione et al. Citation1998]. To the best of our knowledge five genes were disrupted as a result of interruption by an X;autosome translocation near our critical region have been observed. These are XPNPEP2 (MIM *300145) in Xq25, POF1B (MIM *300603) in Xq21.2, DACH2 (MIM *300608) in Xq21.3 [Bione et al. Citation2004], CHM (MIM *300390) in Xq21.2 [Cremers et al. Citation1990], and DIAPH2 (MIM *300108) in Xq22 [Bione et al. Citation1998]. However, most of the breakpoints mapped in the Xq region described in POF patients are intergenic [Mumm et al. Citation2001; Prueitt et al. Citation2002]. Only the DIAPH2 gene was disrupted by translocation in a family with POF affecting oogenesis [Bione et al. Citation1998]. In many cases candidate genes were neither expressed in the ovarian follicle nor in oocyte. It has been suggested that expression of these genes may be altered by a position effect of the balanced translocation [Rizzolio et al. Citation2007]. In our case, the translocation breakpoint falls within the HDX which is the only gene in Xq21 that is expressed in the ovary [EMBL-EBI atlas].
Recent studies have improved knowledge in regard to the X;autosome translocations and POF. Bertini et al. [2010] described an infertile woman with primary amenorrhea due to X;autosome translocation involving Xq22 region. The array CGH results excluded deletions and duplications. Bertini et al. [2010] identified the NFX5 gene which is located on the Xq22.1 region and shows functional homology fragile X mental retardation 1 (FMR1) gene as a POF candidate [Bertini et al. Citation2010]. Giacomozzi et al. [2010] reported an adolescent girl with a de novo unbalanced translocation t(X;15)(q24;q26.3) with partial Xq24 duplication who presented with secondary amenorrhea and absence of pubic and axillary hair. It has been suggested that a distal X breakpoint involving POF1 locus caused POF without virilization during adolescence [Giacomozzi et al. Citation2010].
The disruption of a gene at the translocation breakpoint would be the most probable explanation of the phenotype in female balanced X;autosome translocation patients with a completely skewed XCI pattern. However, other mechanisms may be suggested on the cellular, chromosomal, and transcriptional level. First, defective pairing between the X chromosome and the translocated segment of chromosome 15 within the pachytene oocyte may lead to the elimination of germ cells from the pool [Schlessinger et al. Citation2002]. Second, translocation of regulatory domains to another position on the genome might cause alternations in transcriptional regulation due to ‘position effect’ [Persani et al. Citation2009]. Lastly, in female balanced X;autosome translocations carriers, the normal X chromosome is usually inactivated allowing full expression of genes on the translocated segments [Schmidt and Du Sart Citation1992; Yang et al. Citation2011]. Female balanced X;autosome translocation carriers are generally phenotypically normal because the normal X chromosome is completely inactivated in these patients as a result of phenotypic plasticity to prevent deleterious monosomy. Similarly, as reported here a balanced X;15 translocation has a completely skewed XCI pattern in which the selective pressure for XCI should be against the translocated autosomal material in order to prevent deleterious monosomy.
In this study, we directly demontrated that X;15 translocation interrupts the Xq21 HDX gene in a phenotypically normal female with POF. The findings and our observations suggested that HDX could be critically important in the development of ovarian follicles or the regulation of ovarian gene expression and alterations of this gene may lead to premature ovarian failure. Further characterization and analysis of the genomic region encompassing HDX, in POF patients, as well as its effects on RNA and protein levels in patients should provide information for ovarian failure.
Materials and Methods
Prior to the study, the patient and her family was informed and gave signed informed consent to participate in this study. All protocols involving the use of human tissue specimens were approved by the Institutional Review Board.
Cytogenetic analysis
Chromosomal analysis was performed from peripheral blood samples using standard procedures. G banding analysis was performed on metaphases obtained from PHA stimulated lymphocytes from the proband and all of her family members according to standard procedures.
FISH analysis
FISH was performed on lymphocyte metaphase spreads of the patient using the CEP X SpectrumOrange/Y SpectrumGreen Direct Labeled Fluorescent DNA Probe Kit (Abbott Molecular 07J20-050, Des Plaines, IL) and Whole Chromosome Painting probe 15 (Cytocell, Aquarius®LPP15 G9X, UK).
Cytogenetics array analysis
Molecular cytogenetic array analysis was performed in DNA sample obtained from the patient, according to the manufacturer's protocol for Affymetrix GeneChip® Cytogenetics 2.7M Whole-Genome Array (Affymetrix Inc. Santa Clara, CA, USA) with average genome coverage of approximately one marker per 735 bp. The array consisted of approximately 2.7 million markers, including 2,361,876 non-polymorphic markers and 400,103 single nucleotide polymorphic markers for unbiased coverage of the genome. The microarray image data was extracted using the Command Console Software 3.0.1 (AGCC) (Affymetrix Inc.). Data were analyzed using Chromosome Analysis Suite Software v1.0 (ChAS) (Affymetrix Inc.) with a threshold SNP QC ≥ 1.1 MAPD ≤ 0.27. Duplication or loss of chromosome number at given loci was considered significant only if the duplication or loss of the chromosome exceeded 100 kb. The genomic locations were retrieved from the National Center for Biotechnology Information (NCBI) build 28 (hg 18). The gene located at the translocated X chromosome region is HDX gene and on chromosome 15 includes: LOC727832, LOC727924, CXADRP2, LOC283767, POTEB [MIM 608912], GOLGA8D, LOC646214, LOC650137, BCL8 [MIM 601889], OR4M2, OR4N4, GOLGA8C.
X-chromosome inactivation analysis
DNA was extracted from 200 µl venous blood sample by NucleoSpin Blood kit as in the protocol supplied by the manufacturer (Macherey-Nagel, Düren, GERMANY) and evaluated for quality and concentration using ND-1000 spectrophotometer (Nanodrop, Wilmington, USA) and agarose gel electrophoresis.
Genotyping of the highly polymorphic CAG repeat in the androgen-receptor (AR) was performed to assess the XCI pattern [Allen et al. Citation1992]. The DNA was divided into two identical aliquots, one of which was incubated overnight at 37°C with methylation-sensitive restriction enzyme HpaII (MBI Fermentas, Vilnius, Lithuania) for the digestion of unmethylated (or active) alleles. A second restriction enzyme, RsaI (MBI Fermentas), which recognizes a four base pair sequence not present in the amplified region of the AR locus was also included in the reaction to facilitate the HpaII digestion process. The other aliquot of the DNA was digested with RsaI alone as a control. Male DNA with cytogenetically verified 46,XY karyotype was used as a control for incomplete digestion. After restriction enzyme digest, residual DNA was amplified by using the primers 5'-GTC CAA GAC CTA CCG AGG AG-3' and 5'-CCA GGA CCA GGT AGC CTG TG-3'. PCR products were separated on 8% denaturing 29:1 acrylamide/bisacrylamide gel for 3 h at 15W. Densitometric analysis of the alleles was performed at least twice for each sample using the appropriate software (MultiAnalyst version 1.1; Bio-Rad, Hercules, California). Since the number of cycles could be critically important for the outcome of densitometric analyses, samples were subjected to 30 cycles of amplification during PCR. Products were visualized by ethidium bromide staining, and densitometric analysis of the alleles was performed at least twice for the MultiAnalyst version 1.1software. A corrected ratio (CrR) was calculated by dividing the ratio of the predigested sample (upper/lower allele) by the ratio of the non-predigested sample for normalization of the ratios that were obtained from the densitometric analyses. The use of CrR compensates for preferential amplification of the shorter allele when the number of PCR cycles increases. A skewed population is defined as a cell population with greater than 80% expression of one of the AR alleles. This corresponds to CrR values of < 0.33 or > 3.
Abbreviations
| FISH: | = | fluorescence in situ hybridization |
| XCI: | = | X chromosome inactivation |
| POF: | = | premature ovarian failure |
| POI: | = | premature ovarian insufficiency |
| USG: | = | ultrasonography |
| FSH: | = | follicle-stimulating hormone |
| LH: | = | luteinizing hormone |
| TSH: | = | thyroid-stimulating hormone |
| HDX: | = | highly divergent homeobox |
| AR: | = | androgen-receptor |
| CrR: | = | corrected ratio. |
Acknowledgment
We thank Aysegul Akman, AY-KA Company, for Molecular Cytogenetic Array Analysis.
Declaration of interest: The authors report no conflict of interest.
Author contributions: Cytogenetic analysis and editing of manuscript: GO; Preparation of the manuscript and cytogenetic analysis: SG; XCA and preparation of related parts in manuscript: OEO; FISH analysis: AT; XCA and editing of manuscript:TO; Clinical evaluation of the patient: IK.
References
- Allen, R.C., Zoghbi, H.Y., Moseley, A.B., Rosenblatt, H.M. and Belmont, J.W. (1992) Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 51:1229–1239.
- Beck-Peccoz, P. and Persani, L. (2006) Premature ovarian failure. Orphanet J Rare Dis. Apr 6;1:9.
- Bertini, V., Ghirri, P., Bicocchi, M.P., Simi, P. and Valetto, A. (2010) Molecular cytogenetic definition of a translocation t(X;15) associated with premature ovarian failure. Fertil Steril 94(3):1097.
- Bione, S., Sala, C., Manzini, C., Arrigo, G., Zuffardi, O., Banfi, S., (1998) A human homologue of the Drosophila melanogaster diaphanous gene is disrupted in a patient with premature ovarian failure: evidence for conserved function in oogenesis and implications for human sterility. Am J Hum Genet 62:533–541.
- Bione, S., Rizzolio, F., Sala, C., Ricotti, R., Goegan, M., Manzini, M.C., (2004) Mutation analysis of two candidate genes for premature ovarian failure, DACH2 and POF1B. Hum Reprod 19:2759–2766.
- Cordts, E.B., Christofolini, D.M., Dos Santos, A.A., Bianco, B. and Barbosa, C.P. (2011) Genetic aspects of premature ovarian failure: a literature review. Arch Gynecol Obstet 283:635–643.
- Cremers, F.P., Sankila, E.M., Brunsmann, F., Jay, M., Jay, B., Wright, A., (1990) Deletions in patients with classical choroideremia vary in size from 45 kb to several megabases. Am J Hum Genet 47:622–628.
- Giacomozzi, C., Gullotta, F., Federico, G., Colapietro, I., Nardone, A.M. and Cianfarani, S. (2010) Premature ovarian failure, absence of pubic and axillary hair with de novo 46,X,t(X;15)(q24;q26.3). Am J Med Genet 152A(5):1305–1309.
- Mumm, S., Herrera, L., Waeltz, P.W., Scardovi, A., Nagaraja, R., Esposito, T., (2001) X/autosomal translocations in the Xq critical region associated with premature ovarian failure fall within and outside genes. Genomics 76:30–36.
- Persani, L., Rossetti, R., Cacciatore, C. and Bonomi, M. (2009) Primary Ovarian Insufficiency: X chromosome defects and autoimmunity. J Autoimmun 33:35–41.
- Prueitt, R.L., Chen, H., Barnes, R.I. and Zinn, A.R. (2002) Most X;autosome translocations associated with premature ovarian failure do not interrupt X-linked genes. Cytogenet Genome Res 97:32–38.
- Rizzolio, F., Sala, C., Alboresi, S., Bione, S., Gilli, S., Goegan, M., (2007) Epigenetic control of the critical region for premature ovarian failure on autosomal genes translocated to the X chromosome: a hypothesis. Hum Genet 121:441–450.
- Schlessinger, D., Herrera, L., Crisponi, L., Mumm, S., Percesepe, A., Pellegrini, M., (2002) Genes and translocations involved in POF. Am J Med Genet 111:328–333.
- Schmidt, M. and Du Sart, D. (1992) Functional disomies of the X chromosome influence the cell selection and hence the X inactivation pattern in females with balanced X-autosome translocations: a review of 122 cases. Am J Med Genet 42:161–169.
- Speroff, L. and Fritz, M.A. (2005) Amenorrhea. In: Clinical gynecologic endocrinology and infertility. 7th ed. Lippincott Williams & Wilkins, Philadelphia, Pa, USA 401–464.
- Yang, C., Chapman, A.G., Kelsey, A.D., Minks, J., Cotton, A.M. and Brown, C.J. (2011) X-chromosome inactivation: molecular mechanisms from the human perspective. Hum Genet 130:175–185.