
Abstract
Small supernumerary marker chromosomes (sSMCs) originating from chromosome 10 are rare. A limited number of cases are documented. We report a new diagnosis of a mosaic sSMC (10) in a normal female who asked for genetic evaluation before undergoing controlled ovarian hyperstimulation, in vitro fertilization, and embryo transfer. Chromosome preparations from peripheral lymphocyte cultures were performed according to standard procedures. QFQ-banded chromosomes confirmed the presence of an sSMC: 47,XX,+mar[49]/46,XX[51]. FISH and array CGH analysis showed that the sSMC consisted of chromosome 10 with a gain of the 10p11.1p11.21 (2.5 Mb) chromosomal region. The presence of sSMC (10) was also confirmed in the patient’s mother and sister. It did not appear to affect the phenotype of the women who were phenotypically normal and healthy, and at the time of writing the woman became pregnant naturally. Phenotypes associated with an sSMC vary from normal to severely abnormal. It has been shown that variations in the chromosomal region of sSMCs result in observable differences in clinical outcome. The phenotypical consequences of sSMCs are difficult to predict because of differences in euchromatic DNA content, chromosomal origin, and varying degrees of mosaicism. Therefore, the continued investigation of a larger number of sSMC cases, in particular those originating from chromosome 10 that are the infrequently encountered and characterized, and a better understanding of the genetic content is important in order to improve the delineation of karyotype-phenotype correlation, contributing to a more informed prenatal counseling or prognosis.
Introduction
Small supernumerary marker chromosomes (sSMCs) are structurally abnormal chromosomes which occur in addition to the 46 human chromosomes; they are generally equal in size to, or smaller than, chromosome 20 of the same metaphase spread and they cannot be identified or characterized by conventional banding techniques [Chen et al. Citation2010; Liehr et al., Citation2004a, Citation2004b, Citation2004c]. The risk for phenotypical abnormalities associated with an sSMC depends on several factors, including inheritance, chromosomal origin, morphology, genetic content, presence of uniparental disomy, or degree of mosaicism. In fact, phenotypes associated with an sSMC are variable, ranging from normal to severely affected. It has been shown that, depending on the chromosomal region of an sSMC, differences can be observed and expected in clinical outcome. sSMCs can be detected postnatally or prenatally, the latter being a major problem for cytogenetic investigation and genetic counseling. In a review published by Liehr and Weise [Citation2007], the sSMC frequency was estimated at 0.044% in newborns, 0.075% in prenatal cases, 0.288% in patients with intellectual disability, and 0.122% in infertile patients.
Chromosome 10 is rarely involved in the formation of marker chromosomes, and it represents the most poorly characterized group. Only eight cases have been reported [Blennow and Tillberg Citation1996; Chen et al. Citation2001; Huang et al. Citation2006; Levy et al. Citation2000; Schlegel et al. Citation2002; Snyder et al. Citation1984; Sung et al. Citation2009; Trimborn et al. Citation2005]. In five of these, it was reported that the marker chromosome was associated with an increased risk of abnormal phenotype: severe growth retardation, mental retardation, short stature, several muscle-skeleton deformity, and microcephaly. The identification of the sSMC by conventional cytogenetic analysis alone is almost impossible: in fact modern molecular cytogenetic methods, such as fluorescence in situ hybridization (FISH) and array comparative genomic hybridization (aCGH), are useful and powerful tools to identify the origin of sSMC. Herein, we present the molecular cytogenetic characterization of mosaicism for an sSMC derived from chromosome 10 in a family with apparently normal phenotype, using aCGH and FISH.
Clinical Report
A 40-year old Italian woman came to our attention during the ART workup. She was phenotypically normal, no developmental delay, learning problems, or intellectual disability. Apart from the woman’s long-term infertility, her family history was noncontributory. There were not any known causes of male and female infertility and physical examinations, especially gynecological and urological examinations, were normal. Furthermore, laboratory data showed no biological, hormonal, coagulation, or semen anomalies. All the family members had a normal phenotype, and the family history revealed that the mother had three healthy children, and that the sister of the patient already had a four year old daughter, who was developing normally but we do not have the baby’s cytogenetic information because invasive prenatal diagnostic procedure was not preformed as medical indication was not indicated.
Results
Cytogenetic analyses of cultured lymphocytes of patient using QFQ-banding techniques on 100 metaphases from two colonies documented the presence of an sSMC (47,XX,+mar) in 49% of the lymphocytes examined; the remaining 51% was 46,XX (). A detailed molecular cytogenetic characterization using aCGH was needed to evaluate the size and the genomic constitution of the sSMC with precision. The aCGH analysis demonstrated a 2.5 Mb gain in the gene dosage encompassing the 10p11.21/p11.1 region (36,661,468–39,116,568). FISH studies were performed under stringent conditions using a chromosome 10-specific DNA probe (CEP 10, Cytocell Acquarius, Cambridge, UK) and these showed that the marker had a CEP 10 signal (). No additional hybridization signal was detected on any other chromosomes, eliminating the possibility of an insertion or a translocation elsewhere. These findings confirmed that the marker originated from chromosome 10. Cytogenetic analysis performed from cultured peripheral lymphocytes in the patient’s first-degree relatives, showed the presence of the marker chromosome in her mother (39% normal cells with 61%, sSMC cells, with defined karyotype 47,XX,+mar[61]/46,XX[39]), and in her sister with the sSMS in 77% of the lymphocytes examined, (47,XX,+mar[77]/46,XX[23]), while normal karyotypes (46,XY) were observed in her father and brother. In addition, FISH analysis, using the chromosome 10 CEP specific probe, showed hybridization on both normal chromosomes 10 and on the smaller sSMC. These results thus confirmed that, the marker chromosome was a supernumerary derived from chromosome 10.
Figure 1. Conventional and FISH cytogenetic findings. (a) A QFQ banded metaphase spread showing the supernumerary small marker chromosome (indicated). (b) FISH on metaphase spread using CEP 10 probe (white [green]), showing the both chromosomes 10, and the smaller sSMC. FISH: fluorescence in situ hybridization.
![Figure 1. Conventional and FISH cytogenetic findings. (a) A QFQ banded metaphase spread showing the supernumerary small marker chromosome (indicated). (b) FISH on metaphase spread using CEP 10 probe (white [green]), showing the both chromosomes 10, and the smaller sSMC. FISH: fluorescence in situ hybridization.](/cms/asset/2fddd0bf-3ab6-4ef5-88cf-53148dae6ec8/iaan_a_1067936_f0001_c.jpg)
Discussion
Our report describes a woman with a long-term infertility, presenting an apparently normal phenotype and carrying an sSMC which was identified by QFQ banding analysis. Array CGH, followed by confirmation through FISH experiments, showed that the sSMC derived from chromosome 10 and corresponded to a 2.5 Mb genomic gain, spanning the region 10p11.1p11.21. The presence of the sSMC originating from chromosome 10 was also detected in the woman’s mother and sister; whereas her father and brother had normal karyotypes 46,XY. The region 10p11.1p11.21 detected in the case described contains 3 OMIM genes: the ANKRD30A gene, which has been tested for its association to Alzheimer’s Disease and breast neoplasms and appears to participate in processes associated with the regulation of transcription, as well as the ZNF25 and ZNF33A genes, which seem to be involved in transcriptional regulation. To our knowledge, none of these genes have ever been associated with infertility or pregnancy impairment; further studies are thus needed to understand the effects of the expression of these genes in a triplicate condition, and whether this has a pathogenic role.
In recent years, supernumerary marker chromosomes have been characterized by molecular cytogenetics for their chromosomal origin in over 1,500 patients [Liehr et al. Citation2004a, Citation2004b, Citation2004c]. The effects of a sSMC seem to depend on its origin, size, content and the structure, as well as on the degree of mosaicism, the varying amounts of euchromatin, and their parental origin when the marker contains imprinted genes. The interpretation of the clinical significance of an sSMC is extremely problematic, as sSMC have heterogeneous phenotypical expression [Guediche et al. Citation2012].
Markers derived from the chromosome 10 represent the most poorly characterized group, because it is rarely involved in the formation of marker chromosomes. To date, only eight other cases have been reported: five cases showed abnormal phenotypes and of these, four were extra ring chromosomes; the other three were associated with normal phenotype (). However, it should be taken into account that these three cases, which were reportedly normal at term or during pregnancy, were ascertained at prenatal diagnosis and no information about a long follow-up period is currently available. The present case is the only one which has been identified in adulthood, and cases of intrachromosomal duplication involving the same region have not yet been reported. The approximate breakpoints/genetic content of the sSMC (10) documented with or without clinical findings are shown in . These reported SMCs were de novo in all cases and the degree of mosaicism varied from 35% to 100%. In spite of the same chromosome origin, a great variation of phenotypes was observed among these patients. When a mosaicism occurs, it is very difficult to accurately foresee the phenotype because the mosaicism levels are usually only established from one cell lineage and the levels in the other untested cell lineages may differ. For these reasons, it is difficult to predict the risk associated with an sSMC in the presence of mosaicism.
Figure 2. Cytogenetic and clinical summary of patients with marker chromosome 10 described in the literature. Representation of the approximate breakpoints/genetic content of the reported sSMC(10) cases and of our case with or without clinical symptoms and with a sufficient cytogenetic characterization. Light gray (green) squares correspond to reported sSMC(10) without clinical symptoms and dark gray (red) squares to reported sSMC(10) associated with clinical findings. Breakpoints’ representation is approximated. L: lymphocytes; F: fibroblasts; NEO: neocentromere; AFC: amniotic fluid cells; CVS: chroinic villi sampling; OMC: oral mucosal cells.
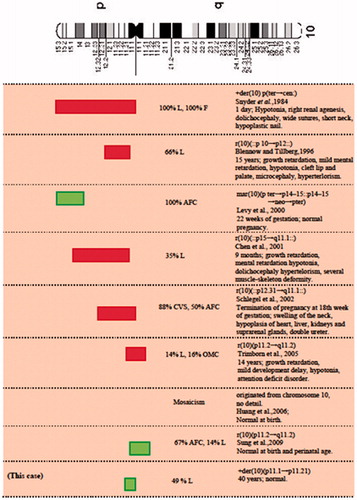
The different phenotypes observed in several patients are also most probably due to different DNA sequences, and thus to the gene content of the sSMC. Most sSMCs are ascertained in phenotypically abnormal subjects, but it is not always possible to correlate the phenotype with the presence of the marker chromosome. In the case in question, the presence of the sSMC does not correlate with an abnormal phenotype and this is probably due to the minimal involved material of chromosome 10 and thus gene content of the sSMC. In cases like this, with apparently normal phenotypes, the characterization of an sSMC can also provide important information about the regions that are phenotypically silent in the presence of imbalanced gene dosage [Liehr et al. Citation2006; Starke et al. Citation2003; Sumption and Barber Citation2001].
Moreover, we wish to underline that it has been reported that the frequency of sSMCs detected in infertile patients is higher than that in the general population (between 0.122% and 0.125% versus 0.044%) and it is also different between male (0.165%) and female infertility (0.022%) [Guediche et al. Citation2012; Liehr and Weise Citation2007]. In our case, the presence of the sSMC did not give rise to reproductive disorders in the patient’s mother (who had three sons) and her sister (one daughter). Furthermore, at the time of writing, both the patient and her sister became pregnant naturally and their pregnancies are proceeding normally, without any detected abnormalities in fetuses but both at this time are just a week shy of chorion villi (CVS) or amniotic fluid (AFS) assessment. Nevertheless, since the mosaicism levels of sSMC may be very different in different tissues, it cannot be definitively excluded that the sSMC may contribute to the infertility. Thus, whether the ch.10-derived sSMC is a cause or a coincidental finding is still questionable since the mechanism by which sSMC influence fertility has not yet been understood [Manvelyan et al. Citation2008].
In conclusion, this report highlights the difficulty in ascertaining a precise phenotype-karyotype correlation and phenotypic outcome, mainly due to breakpoint distribution heterogeneity and to different levels and distribution of mosaicism. We wish to stress and clarify the need to use specific techniques like FISH or aCGH to obtain more accurate knowledge about the size of the segment causing a partial trisomy and its gene content. This would greatly improve the ability to predict phenotype and contribute to better-informed prenatal counseling and prognosis. Indeed, this kind of study could give detailed information about disease-related genes, where increasing gene dosage might play an important role. The present work shows the importance of studying patients who have comparable chromosome defects, to identify clinical similarities so as to benefit the counseling of future cases.
Materials and Methods
Routine cytogenetics were performed from cultured peripheral lymphocytes with QFQ-banding using standard procedures (Q-banding obtained by Quinacrine). FISH analyses were performed on lymphocyte metaphase spreads of the woman, under stringent conditions using a chromosome 10-specific DNA probe (CEP 10, Cytocell Acquarius, Cambridge, UK). Genomic imbalances were analyzed by aCGH according to the manufacturer’s recommended protocol, using a Kit Cytochip oligo ISCA 4x180K (BlueGnome, an Illumina company, Fulbourn, Cambridge, UK) with a median of around 100 kb resolution; for the genomic positions we used the version 19 of the Human Genome (http://genome.ucsc.edu/). Further, standard chromosome analysis from cultured peripheral lymphocytes in the patient’s first-degree relatives (mother, father, sister, and brother) was carried out.
Declaration of interest
The authors report no conflict of interest.
Author contributions
Performed experiments, prepared the figures, and contributed to the analysis of the results: AP, MG; Recruited participants, collected and interpreted the data: AL; Interpreted the data, wrote the draft and final manuscript: RS, RT; Interpreted the data, and revised the draft and final manuscript: MM. All authors approved revisions of the final manuscript.
References
- Blennow, E. and Tillberg, E. (1996) Small extra ring chromosome derived from chromosome 10p: clinical report and characterization by FISH. J Med Genet 33:399–402
- Chen, C.P., Chen, M., Ko, T.M., Ma, G.C., Tsai, F.J., Tsai, M.S., et al. (2010) Prenatal diagnosis and molecular cytogenetic characterization of a small supernumerary marker chromosome derived from chromosome 8. Taiwan J Obstet Gynecol 49:500–505
- Chen, Z., Meloni-Erig, A., Palumbos, J.C., Guan, X.Y., Carroll, K.L., Dent, K.M., et al. (2001) Pure tirsomy 10p resulting from an extra ring chromosome: Characterization by methods of advanced molecular cytogenetics. Am J Med Genet 102:379–382
- Guediche, N., Tosca, L. Nouchy, M., Lecerf, L., Cornet, D., Brisset, S., et al. (2012) Small supernumerary marker chromosomes derived from chromosomes 6 and 20 in a woman with recurrent spontaneous abortions. Eur J Med Genet 55:737–742
- Huang, B., Solomon, S., Thangavelu, M., Peters, K. and Bhatt, S. (2006) Supernumeraray marker chromosomes detected in 100000 prenatal diagnoses: Molecular cytogenetic studies and clinical significance. Prenat Diagn 26:1142–1150
- Levy, B., Papenhausen, P., Tepperberg, J., Dune, T., Fallet, S., Magid, M., et al. (2000) Prenatal molecular cytogenetic diagnosis of partial tetrasomy 10p due to neocentromere formation in an inversion duplication analphoid marker chromosome. Cytogenet Cell Genet 91:165–170
- Liehr, T., Claussen, U. and Starke. H. (2004a) Small supernumerary marker chromosomes (sSMC) in humans. Cytogenet Genome Res 107:55–67
- Liehr, T., Hickmann, G., Kozlowski, P., Claussen, U. and Starke, H. (2004b) Molecular-cytogenetic characterization of the origin and the presence of pericentromeric euchromatin on minute supernumerary marker chromosomes (SMCs). Chromosome Res 12:239–244
- Liehr, T., Mrasek, K., Weise, A., Dufke, A., Rodriguez, L., Martinez Guardia, N., et al. (2006) Small supernumerary marker chromosomes:–progress towards a genotype-phenotype correlation. Cytogenet Genome Res 112:23–34
- Liehr, T., Mrasek, K., Weise, A., Kuechler, A., von Eggeling, F., Claussen, U., et al. (2004c) Characterization of small supernumerary marker chromosomes (sSMC) in human. Current Genomics 5:279–28
- Liehr, T. and Weise, A. (2007) Frequency of small supernumerary marker chromosomes in prenatal, newborn, developmentally retarded and infertility diagnostics. Int J Mol Med 19:719–731
- Manvelyan, M., Riegel, M., Santos, M., Fuster, C., Pellestor, F., Mazaurik, M. L., et al. (2008) Thirty-two new cases with small supernumerary marker chromosomes detected in connection with fertility problems: detailed molecular cytogenetic characterization and review of the literature. Int J Mol Med 21:705–714
- Schlegel, M., Baumer, A., Riegel, M., Wiedemann, U. and Schinzel, A. (2002) Maternal uniparental isodisomy 10 and mosaicism for an additional marker chromosome derived from the paternal chromosome 10 in a fetus. Prenat Diagn 22:418–421
- Snyder, F.F., Lin, C.C., Rudd, N.L., Shearer, J.E., Heikkila, E.M. and Ho, J.J. (1984) A de novo case of trisomy 10p: Gene dosage studies of hexokinase, inorganic pyrophosphatase and adenosine kinase. Hum Genet 67:187–189
- Starke, H., Nietzel, A., Weise, A., Heller, A., Marsek, K., Belitz, B., et al. (2003) Small supernumerary marker chromosomes (SMCs): Genotype-phenotype correlation classification. Hum Genet 114:51–67
- Sumption, N.D. and Barber, JC. (2001) A transmitted deletion of 2q13 to 2q14.1 causes no phenotypic abnormalities. J Med Genet 38:125–127
- Sung, P.L., Chang, S.P., Wen, K.C., Chang, C.M., Yang, M.J., Chen, L.C., et al. (2009) Small supernumerary marker chromosome originating from chromosome 10 associated with an apparently normal phenotype. Am J Med Genet Part A 149A:2768–2774
- Trimborn, M., Grueters, A., Neitzel, H. and Tönnies, H. (2005) First small supernumerary ring chromosome carrying 10q euchromatin in a patient with mild phenotype characterized by molecular cytogenetic techniques and review of the literature. Cytogenet Genome Res 108:278–282