Abstract
Background:
Dalfampridine extended release tablets (dalfampridine-ER, known as prolonged-, modified, or sustained-release fampridine tablets in some countries) are approved for the improvement of walking in patients with multiple sclerosis (MS). Dalfampridine-ER is an extended release formulation of 4-aminopyridine (4-AP). Dalfampridine-ER is incorporated into MS management strategies that may include disease-modifying and symptomatic therapies. Since several symptomatic therapies are partially or fully metabolized by enzymes of the hepatic cytochrome P450 system (CYP450) it is important to evaluate drug–drug interactions through potential effects of dalfampridine-ER on CYP450.
Methods:
The ability of 4-AP to inhibit CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4/5 in a direct and time-dependent manner was evaluated using pooled human liver microsomes. 4-AP concentrations were 0.03, 0.1, 0.3, 1, 3, 10, and 30 μM, representing 0.1–100-times the average plasma 4-AP concentration (30 ng/mL; 0.32 μM) at therapeutic dosing; the concentration inhibiting 50% of each enzyme activity (IC50) was determined. The ability of 4-AP (0.025, 0.25, 2.5, and 25 μM) to induce the expression of CYP1A2, 2B6, 2C9, 2C19, 2E1, and 3A4/5 enzymes was evaluated using primary cultures of freshly isolated human hepatocytes from non-transplantable livers. The enzyme-inducing effects of 4-AP were compared with the prototypical inducers. Metabolites were assayed using high-performance liquid chromatography-tandem mass spectrometry techniques. All inhibition and induction assays included positive controls.
Results:
4-AP did not directly inhibit CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4/5, but at a concentration of 30 μM, CYP2E1 was inhibited by 12%, resulting in an estimated IC50 value of 125 μM. None of the enzymes demonstrated time-dependent inhibition by 4-AP. There was little or no effect by 4-AP on enzyme induction, with enzyme activities approximately equivalent to vehicle control. A main limitation was the inability to estimate effectiveness of 4-AP relative to prototypical CYP450 inducers.
Conclusion:
The likelihood of drug–drug interactions is remote in patients with MS who may be taking dalfampridine-ER concomitantly with medications that are metabolized by CYP450 pathways.
Background
Dalfampridine extended release tablets (dalfampridine-ER, known as prolonged-, modified, or sustained-release fampridine tablets in some countries), are approved for the improvement of walking in patients with multiple sclerosis (MS)Citation1. This approval was based on two Phase 3 clinical trials that demonstrated a significant improvement in walking speed relative to placebo among patients who responded to treatmentCitation2,Citation3.
Dalfampridine-ER is an extended release formulation of dalfampridine or 4-aminopyridine (4-AP), a potassium channel blocker that has been extensively used as a tool for characterizing potassium channels. Its putative mechanism of action in MS is relief of the conduction block in demyelinated axonsCitation4, although it may also enhance neurotransmission at synapsesCitation5,Citation6. Since this drug is incorporated into MS management strategies that may include a number of other drugs, including disease-modifying therapies (DMTs), it is important to consider its potential for drug–drug interactions.
Many drug–drug interactions are mediated by the hepatic cytochrome P450 system (CYP450). Interferons have been suggested to impact the CYP450 system, thereby potentially altering the pharmacokinetics and pharmacodynamic effects of concomitant medicationsCitation7. However, the CYP450 system itself is not involved in the metabolism of traditional, injectable DMTs for MS, such as the interferons, glatiramer acetate, and natalizumab, and 4-AP itself is largely excreted unchanged in the urine (discussed below), suggesting low potential risk for drug interactions with concomitant use of dalfampridine-ER. Indeed, in a sub-group analysis of the clinical trials, the tolerability and efficacy of dalfampridine-ER appeared to be similar in patients using and not using these therapiesCitation8. Those results suggested that dalfampridine-ER can be effective regardless of DMT use, and is likely to have a safety profile in patients using DMTs that is similar to those not using them. However, in contrast to DMTs, several pharmacologic therapies used for the symptomatic treatment of MS are partially or fully metabolized by enzymes of the CYP450 system, including baclofen and tizanidine for spasticity, tolterodine for neurogenic bladder, dextromethorphan/quinidine for pseudobulbar affect, and a variety of antidepressants that may be used for depression or neuropathic pain. In addition to these treatments related to MS, there may be a variety of other drugs prescribed for concurrent conditions in individual patients.
The pharmacokinetics of 4-AP have been evaluated in several studiesCitation9–13, which have included the demonstration that it is rapidly and completely excreted by the urinary route, predominantly as an unchanged compoundCitation11. Excretion of unchanged drug suggests that 4-AP does not undergo extensive hepatic metabolism and is therefore not an important substrate of the CYP450 system. However, drug–drug interactions might nevertheless be possible if 4-AP were to inhibit or induce CYP450 enzymes involved in the metabolism of other drugs. Therefore, the purpose of this in vitro study was to determine whether 4-AP has an effect, either by induction or inhibition, on components of the CYP450 system that could potentially result in drug interactions. The study was performed in accordance with US Food and Drug Administration (FDA) guidance for performing drug interaction studiesCitation14, and includes evaluation of the clinically relevant enzymes CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4/5.
Methods
Chemicals and reagents
The 4-AP was produced by Regis Technologies (Morton Grove, IL). Stock and working solutions of 4-AP were prepared fresh daily in high-purity water (for inhibition studies) or dimethyl sulfoxide (DMSO).
The following reagents were purchased from Sigma-Aldrich (St. Louis, MO): acetaminophen, 3-amino-1,2,4-triazole, ammonium acetate, bupropion HCl, chlorzoxazone, coumarin, dextromethorphan, dextrorphan, diclofenac, DMSO, furafylline, glucose-6-phosphate, glucose-6-phosphate dehydrogenase, ethylenediaminetetraacetic acid, tetrasodium salt (EDTA), 6-hydroxychlorzoxazone, 7 hydroxycoumarin (umbelliferone), 4′-hydroxydiclofenac, (±)-4′-hydroxymephenytoin, 6β-hydroxytestosterone, ketoconazole, magnesium chloride, 8-methoxypsoralen, 4 methylpyrazole, midazolam, modafinil, α-naphtho-flavone, β-nicotinamide-adenine dinucleotide phosphate (NADP), nicotine, orphenadrine, paroxetine, phenacetin, phencyclidine, quercetin, quinidine, sucrose, sulfaphenazole, testosterone, ticlopidine, Trizma® base and troleandomycin.
Matrigel™ and ITS+ were purchased from BD Biosciences (Bedford, MA). Formic acid was purchased from EMD Chemicals (Gibbstown, NJ). Acetone, acetonitrile, ethanol, hydrochloric acid, methanol, perchloric acid, potassium chloride, potassium hydroxide, sodium chloride and sodium hydroxide were purchased from Fisher Scientific (Pittsburgh, PA). Dulbecco’s Modified Eagle’s Medium (DMEM), GlutaMAX-1, insulin, MEM-non-essential amino acids, modified Eagle’s Medium Dr. Chee’s modification (MCM), and penicillin-streptomycin were purchased from Invitrogen (Grand Island, NY). Carbon dioxide was purchased from Helget Gas (Kansas City, MO). PureCol was purchased from Inamed BioMaterials (Fremont, CA). Fetal bovine serum (FBS) was purchased from SAFC Biosciences (Lenexa, KS). Disodium hydrogen phosphate, potassium dihydrogen phosphate, potassium hydrogen phosphate, sodium hydrogen phosphate, and dipotassium hydrogen phosphate were purchased from Mallinckrodt Baker (Phillipsburg, NJ). Loctite 4013 was purchased from the Loctite Corporation (Rocky Hill, CT). BCA (bicinchoninic acid) Protein Assay Kit was purchased from Pierce Chemical Co. (Rockford, IL). Hydroxybupropion was purchased from BD Gentest (Woburn, MA). 1′ Hydroxymidazolam was purchased from Cerilliant (Round Rock, TX). Tienilic acid was purchased from Cypex Ltd. (Dundee, Scotland). Formic acid was purchased from EMD Chemicals (Gibbstown, NJ). Acetonitrile, acetone, methanol, potassium hydroxide, and sodium hydroxide were purchased from Fisher Scientific (Pittsburgh, PA). Dipotassium hydrogen phosphate and potassium dihydrogen phosphate were purchased from Mallinckrodt Baker (Phillipsburg, NJ). Amodiaquine and N-desethylamodiaquine were purchased from LGC Standards (Teddington, Middlesex, UK). Montelukast was purchased from Sequoia Research Products (Pangbourne, UK). S-Mephenytoin was purchased from Toronto Research Chemicals Inc. (North York, Ontario, Canada). High purity water and gemfibrozil glucuronide were prepared at XenoTech, LLC (Lenexa, KS). 17β-N,N-Diethylcarbamoyl-4-methyl-3-oto-4-aza-5α-androstane-17α-carboxamide (4-MA) was a generous gift from Dr G. H. Rasmusson (Merck Sharp & Dohme, Rahway, NJ).
The deuterated internal standards used were d4-acetaminophen, d5-N-desethylamodiaquine, d3-dextrorphan, d6-hydroxybupropion, d2-6 hydroxychlorzoxazone, d5-7-hydroxycoumarin, d4-4′-hydroxydiclofenac, d3-4′-hydroxymephenytoin, d3-1′-hydroxymidazolam, and d3-6β-hydroxytestosterone. The sources of these standards are not provided due to the proprietary nature of this information.
In vitro CYP450 inhibition
The ability of 4-AP to inhibit the drug-metabolizing enzymes CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4/5 in a direct and time-dependent manner was evaluated, as previously establishedCitation15,Citation16, using human liver microsomes pooled from 16 samples that were prepared and characterized at XenoTech, LLC (Lenexa, KS). In brief, duplicate incubations were conducted at 37 ± 1°C in 400 μL incubation mixtures containing potassium phosphate buffer (50 mM, pH 7.4), MgCl2 (3 mM), EDTA (1 mM, pH 7.4), and P450 marker substrates at concentrations approximately equal to their Km (). For CYP3A4/5 inhibition, two substrates were used (testosterone and midazolam) as recommended by the FDACitation14. Reactions were initiated by addition of an NADPH-generating system containing NADP (1 mM), glucose-6-phosphate (5 mM), and glucose-6-phosphate dehydrogenase (1 Unit/mL). After 5 min, the reactions were terminated by addition of an equal volume of acetonitrile (v/v) containing an appropriate internal standard (). Precipitated protein was removed by centrifugation (920 g, 10 min, 10°C). Calibration and quality control metabolite standards were prepared in zero time incubations.
Table 1. Experimental conditions for measuring inhibition of microsomal CYP450 activity by 4-aminopyridine.
The concentrations of 4-AP that were evaluated (0.03, 0.1, 0.3, 1, 3, 10, and 30 μM) are ∼0.1–100-times the average plasma 4-AP concentration (30 ng/mL; 0.32 μM) that was measured in a clinical trial of the therapeutic dose of dalfampridine-ER (10 mg administered twice daily)Citation2. Because of the possibility that 4-AP may bind to microsomal protein or lipids, an attempt was made to use standard conditions of 0.1 mg/mL microsomal protein, 5 min incubation time, and 50 mM phosphate buffer concentration, for all reactions with the exceptions of coumarin 7-hydroxylation and midazolam 1′-hydroxylation assays. For these reactions, slightly different protein concentrations were used () to allow the rate of reaction to be measured under initial rate conditions.
To evaluate 4-AP as a direct-acting inhibitor, the pooled microsomes were incubated with the marker substrates in the presence and absence of the range of 4-AP concentrations to determine the concentration inhibiting 50% of enzyme activity (IC50) value. For time-dependent inhibition, the same concentrations of 4-AP were pre-incubated with the microsomes and an NADPH-generating system for 30 min. After pre-incubation, the marker substrate was added, and the incubation was continued for 5 min to measure residual P450 activity. All reactions were terminated as described above. Known direct-acting and metabolism-dependent inhibitors were included as positive controls ()Citation14. Samples were analyzed as described in the Analytical methods section.
Table 2. Positive controls for IC50 determinations for inhibition of microsomal CYP450 enzyme activity.
In vitro CYP450 induction
Evaluation of 4-AP as an inducer of the expression of CYP1A2, 2B6, 2C9, 2C19, 2E1, and 3A4/5 enzymes was performed using primary cultures of freshly isolated human hepatocytes with a Matrigel overlay. Of note, CYP2D6 was not examined because this enzyme is recognized by the FDA as being non-inducibleCitation14. Hepatocytes were obtained from non-transplantable livers from three individual donors according to previously described methodsCitation17. Culture and treatment procedures were performed as described by Madan et al.Citation18. The viability of each preparation was analyzed by trypan blue exclusion, and cultures were allowed to adapt for 3 days prior to treatment.
Hepatocytes were exposed to test articles for 3 consecutive days. Each test article was evaluated in each liver hepatocyte preparation. The test articles included vehicle controls of 0.1% DMSO, 0.1% saline, 4-aminopyridine at concentrations of 0.025, 0.25, 2.5, and 25 μM, and prototypical inducers of human CYP450 including omeprazole (100 μM; CYP1A2), phenobarbital (750 μM; CYP2B6), isoniazid (100 μM; CYP2E1), and rifampin (10 μM; CYP2C9, CYP2C19, and CYP3A4/5); DMSO was the vehicle for 4-aminopyridine, omeprazole, phenobarbital, and rifampin, and saline was the vehicle for isoniazid. The concentrations of 4-AP are ∼0.08–80-times the average plasma concentration, 30 ng/mL (0.32 μM), that was determined in a clinical trial of the therapeutic dose of 10 mg twice dailyCitation2.
All working solutions were prepared fresh daily prior to hepatocyte treatment. Approximately 24 h following the final treatment, cultures were visualized to confirm morphological integrity prior to isolation of microsomes, which was carried out according to the methods described by Madan et al.Citation18.
Microsomal protein concentrations were determined using a BCA Protein Assay Kit (Pierce Chemical Company, Rockford, IL) and a Synergy HT Multi-Detection Microplate Reader (BioTek Instruments, Inc., Winooski, VT). Microsomal incubations were conducted in duplicate at 37 ± 1°C in 200 μL volumes of incubation buffer (pH 7.4) consisting of high purity water, potassium phosphate (50 mM), MgCl2 (3 mM), EDTA (1 mM) and the marker substrates, as shown in . The testosterone incubations also contained 17β-N,N diethylcarbamoyl-4-methyl-3-oto-4-aza-5α-androstane-17β-carboxamide (1 mM) in acetone (0.1% v/v) to inhibit steroid 5α reductaseCitation19. These preparations were loaded onto a Tecan Liquid Handling System (Tecan, Research Triangle Park, NC), and reactions were initiated by addition of an NADPH-generating system containing NADP (1 mM), glucose-6-phosphate (5 mM), and glucose-6-phosphate dehydrogenase (1 Unit/mL). Incubation times were either 10 min (CYP2C9, 2E1, and 3A4/5) or 30 min (CYP1A2, 2B6, and 2C19), and reactions were terminated by addition of acetonitrile containing the appropriate internal standards (). Precipitated protein was removed by centrifugation at 920 g (10 min, 10°C), and supernatant fractions were analyzed as described in the Analytical methods section.
Table 3. Experimental conditions for measuring induction of microsomal CYP450 enzyme activity by 4-aminopyridine.
Analytical methods
High-performance liquid chromatography-tandem mass spectrometry (HPLC/MS/MS) was performed using proprietary validated methods for the determination of each metabolite; authentic, deuterated metabolite standards were used in all assays and zero-time incubations served as blanks. Mass spectrometry equipment was either an ABI Sciex (Applied Biosystems/MDS SCIEX, Foster City, CA) API 4000, API 3000, or API 2000 instrument with Shimadzu HPLC pumps and autosampler systems. The ionization mode and mass transitions that were monitored are shown in and for inhibition and induction, respectively. The instruments were equipped with an electrospray (TurboIonSpray) ionization source (Applied Biosystems, Foster City, CA) and two LC-10ADvp pumps with an SIL-HTa autosampler and a DGU-14 solvent degasser (Shimadzu Scientific Instruments, Columbia, MD). In the inhibition studies, the HPLC columns included a Waters Atlantis C18 (5 -μm particle size, 50 mm × 2.1 mm) (Waters, Milford, MA) for the analysis of phenacetin O-deethylation, coumarin 7-hydroxylation, amodiaquine N-dealkylation, diclofenac 4′-hydroxylation, S-mephenytoin 4′-hydroxylation, dextromethorphan O-demethylation, chlorzoxazone 6-hydroxylation, testosterone 6β-hydroxylation, and midazolam 1′-hydroxylation; and a Waters Atlantis T3 (3 -μm particle size, 50 mm × 2.1 mm) (Waters) for bupropion hydroxylation. Analyses of the induction samples included use of a 5 -μm, 100 mm × 2.1 mm, C18 Waters Atlantis column (Waters) for the analysis of phenacetin O-dealkylation; a 5-μm, 50 mm × 2.1 mm, C18 Waters Atlantis column (Waters) for bupropion hydroxylation; a 3-μm, 50 mm × 2.1 mm, T3 Waters Atlantis column (Waters) for diclofenac 4′-hydroxylation, chlorzoxazone 6-hydroxylation, S-mephenytoin 4′-hydroxylation, and testosterone 6β-hydroxylation. All columns were preceded by a direct connection guard column with a C8, 4.0 mm × 2.0 mm cartridge (Phenomenex, Torrance, CA) and were maintained at ambient temperature.
Metabolites were quantified by back-calculation of a weighted (1/x), linear, least-squares regression. The regression fit was based on analyte/internal standard peak-area ratios calculated from calibration standard samples, which were prepared from authentic metabolite standards. Peak areas were integrated with Applied Biosystems/MDS SCIEX (Foster City, CA).
Statistical analyses
For inhibition studies, the IC50 data were processed with a validated, custom software program (DI IC50 LCMS Template version 2.0.3) for Microsoft Excel (Office 2000 version 9.0, Microsoft Inc., Redmond, WA). When inhibition of enzyme activity was observed, the data were processed for determination of IC50 values by non-linear regression with XLfit3 (Version 3.0.5, ID Business Solutions Ltd., Guildford, Surrey, UK). This software uses the Levenberg-Marquardt algorithm for non-linear regression fitting of the data to the following 4-parameter sigmoidal-logistic IC50 equation:
Background was set to 0 with a range up to 100 to express data as a percentage of control. This software has been verified for its ability to calculate IC50 values that lie within the concentration range of 4-AP. When less than 50% inhibition is observed, the data are not extrapolated; hence, IC50 values are reported as being greater than the highest concentration of 4-AP tested.
For the induction studies, data were processed using the validated, custom software program (EI Interim Data Engine, version 1.2.1) for Microsoft Office Excel 2003 (version 11.0, Microsoft Corporation, Redmond, WA). The individual rates of reaction from replicate samples were averaged, and for those groups with n ≥ 3, standard deviations were determined. Statistically significant differences between group means were calculated by equal variance and normality tests to determine if the data were parametrically distributed. For parametrically distributed data sets, with the exception of isoniazid and saline treatment groups, a one-way repeated measures analysis of variance (ANOVA) was carried out to test for significant differences between group means. For isoniazid and saline, a Student’s t-test was used to test for differences. For non-parametrically distributed data sets, with the exception of isoniazid and saline treatment groups, a Kruskal-Wallis ANOVA was performed. The ANOVA was followed by a Dunnett’s post-hoc test to identify the group means that were significantly different from the controls. Statistical analyses were performed with Sigma Stat Statistical Analysis System (version 2.03, Systat Software, Inc., Point Richmond, CA). For all analyses, a p-value <0.05 was considered to indicate significance. Data were graphed with a validated, custom software program (EI Interim Data Engine, version 1.2.1) for Microsoft Office Excel 2003 (version 11.0, Microsoft Corporation, Redmond, WA). The -fold increase was determined by the enzyme rate for each positive test article divided by the control rate. The enzyme-inducing effects of 4-AP were compared with the prototypical inducers in terms of relative effectiveness, which was calculated by the equation:
Guidance by the FDA on drug interaction studies suggests that a drug with a change in enzyme activity that results in effectiveness ≥40% relative to the positive control can be considered an enzyme inducerCitation14.
Results
In vitro CYP450 inhibition
Under the experimental conditions used, there was no direct inhibition of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4/5 at 4-AP concentrations up to 30 μM ( and ). However, at a 4-AP concentration of 30 μM, there was ∼12% inhibition of CYP2E1, resulting in an estimated IC50 value of 125 μM and a Ki for the 4-AP-mediated inhibition of CYP2E1 of 62.5 μM; both of which exceed the highest concentration tested.
Figure 1. Inhibitory effects of 4-aminopyridine on cytochrome P450 enzymes with and without a 30-min pre-incubation with NADPH-fortified human liver microsomes. Substrate concentrations were approximately equal to their Km.
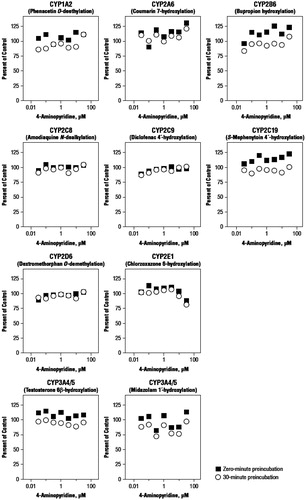
Table 4. In vitro effects of 4-aminopyridine on inhibition of human CYP enzymes.
When 4-AP was pre-incubated for 30 min in the presence of NADPH-fortified human liver microsomes, there was little or no evidence of time-dependent inhibition for any of the enzymes ( and ). Relative to direct inhibition, small increases in inhibition were observed with pre-incubation for CYP1A2, 2A6, 2B6, and C19, although none of these changes was substantial CYP2E1, which had the greatest time-dependent inhibition, and was only inhibited by 19% at 30 μM of 4-AP.
In vitro CYP450 induction
Viability of the hepatocyte preparations was between 83.6% and 91.1%, and these values were considered acceptable for the assays. During and after adaptation to culture, the hepatocytes were judged to be morphologically normal with adequate confluency for treatment. Evaluation of the morphological integrity after final treatment showed that the hepatocytes did not demonstrate any overt signs of toxicity resulting from the test articles.
The mean microsomal CYP enzyme activities after hepatocyte treatment with test articles are summarized in . As shown in , which expresses these activities as the relative increase compared with the vehicle control, hepatocytes responded as expected to treatment with prototypical CYP450 inducers. Treatment with the known inducers resulted in a 35.6-fold increase in activity of CYP1A2 with omeprazole; a 10.7-fold increase in CYP2B6 with phenobarbital; a significant (p < 0.05) 2.6-fold increase in CYP2E1 with isoniazid; significant (p < 0.05) increases in CYP2C9 (1.9-fold) and CYP3A4/5 (6.6-fold) with rifampin; and a 4.2-fold increase in CYP2C19 with rifampin. For CYP1A2 and CYP2B6, although significance was determined among all the treatment groups as a result of Kruskal-Wallis One-way ANOVA on ranks (p < 0.05), Dunnett’s test was unable to specify which individual groups were significantly different.
Figure 2. Change in microsomal cytochrome P450 enzyme activity relative to vehicle control after exposure of cultured human hepatocytes to 4-aminopyridine and prototypical inducers relative to vehicle control. Values are presented as -fold increase over vehicle control based on the absolute values shown in , and are for three determinations (one from each microsomal preparation) unless otherwise indicated.
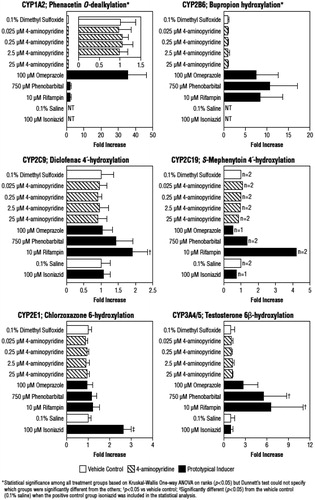
Table 5. Microsomal cytochrome P450 enzyme activity after exposure of cultured human hepatocytes to 4-aminopyridine and prototypical inducers.
In contrast to the known inducers, 4-AP demonstrated little or no effect on enzymes. Enzyme activities in the presence of 4-AP were approximately equivalent to the DMSO vehicle control, with a very narrow range of increases, 0.89–1.2-fold, across concentrations and enzymes ().
Relative effectiveness could not be calculated for 4-AP for CYP2B6 and 2C9, and for three of the four 4-AP concentrations for 2C19 because individual values included negative numbers that precluded determination of an average value (). However, for CYP1A2 and 3A4/5, as well as 2C19 (2.5 μM concentration only), none of the effectiveness values exceeded the FDA’s cut-off of 40% (); the highest effectiveness value was 3.29 ± 5.70% for the 2.5 μM concentration for CYP3A4/5. It should also be noted that, since 4-AP was dissolved in DMSO and the prototypical CYP2E1 inducer (isoniazid) was dissolved in saline, the potential of 4-AP to induce CYP2E1 activity could not be evaluated based on effectiveness.
Table 6. Relative effectiveness of 4-aminopyridine compared with the prototypical inducers (positive controls expressed as 100% for their respective enzymes). Percent relative effectiveness was calculated as described in Methods.
Discussion
Although 4-AP is primarily excreted in the urine as an unchanged drug and is thus not significantly metabolized by CYP enzymesCitation11,Citation20,Citation21, it is important to determine if 4-AP may induce or inhibit CYP activity to further predict if drug–drug interactions may occur with the other drugs regularly used by MS patients. The current analyses confirm that, at therapeutic plasma concentrations of 4-AP, there is little potential of drug–drug interactions through this mechanism. In particular, there were no effects on CYP3A4, which is the most common CYP450 isozyme involved in drug metabolism, and little or no evidence of effects on other evaluated isozymes at 4-AP concentrations that were up to 100-times the mean therapeutic plasma concentration.
While 4-AP did not demonstrate direct inhibitory activity for most enzymes evaluated, there was evidence of low direct inhibition, ∼12%, of CYP2E1 at 30 μM. However, this inhibition was only observed at the highest 4-AP concentration, 30 μM, which is ∼100-times higher than the average plasma 4-AP concentration of 0.32 μM at the therapeutic dose of dalfampridine-ERCitation2, and resulted in an estimated IC50 for CYP2E1 of 125 μM. Furthermore, based upon FDA draft guidanceCitation14, a ratio of the [I]/Ki of at least 0.1, where [I] is the concentration of inhibitor exposed to the active site of the enzyme, warrants clinical drug interaction studies. Since [I] would operationally translate to the Cmax (peak plasma concentration), the resulting ratio for 4-AP would be 0.01, suggesting additional drug interaction studies are not warranted based on the estimated Ki of 62.5 μM for CYP2E1, and a maximum individual Cmax of 67 ng/mL (0.71 μM) that has been reported with therapeutic doses of dalfampridine-ER in a clinical trialCitation2. It should be noted that, although CYP2E1 has been shown to be the predominant enzyme responsible for 3-hydroxylation of 4-AP, the parent compound undergoes very limited metabolic processingCitation22, with clearance primarily as unchanged parent compound in the urineCitation11.
For both CYP2E1 and several other cytochromes, pre-incubation appeared to have a slight impact on inhibition, although there was no substantial increase in CYP2E1 or other cytochrome inhibition relative to the direct assay. However, it is possible that, with longer incubation, a greater inhibitory effect might be observed.
In the induction assays, treatment of three preparations of cultured human hepatocytes with 4-AP at concentrations up to 80-times the therapeutic plasma concentration had little or no effect on CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2E1, or CYP3A4/5 activity. FDA guidance suggests that a drug that produces a change ≥40% of the positive control can be considered as an enzyme inducer and warrants in vivo evaluation; 4-AP had almost no effect on CYP activity for those values that could be calculated. While 4-AP effectiveness values could not adequately be estimated for CYP2B6, 2C9, 2E1, and 2C19, it may nevertheless be considered unlikely that 4-AP would have a clinically significant impact on the induction of these enzymes since there was no increase in activity with 4-AP relative to the vehicle control, whereas hepatocytes responded as expected to treatment with positive controls.
Conclusions
This in vitro study demonstrated that 4-AP, at concentrations up to 100-fold higher than mean therapeutic plasma concentrations, neither inhibits CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4/5 nor induces 1A2, 2B6, 2C9, 2C19, 2E1, and 3A4/5 to a clinically relevant extent. These data suggest that the likelihood of drug–drug interactions is remote in patients with MS who may be taking dalfampridine-ER concomitantly with medications that are metabolized by these pathways.
Transparency
Declaration of funding
This study was funded by Acorda Therapeutics, Inc., Ardsley, New York.
Declaration of financial/other relationship
AC and AB are employees and stockholders of Acorda Therapeutics, Inc., Ardsley, New York.
Acknowledgments
The authors wish to thank E. J. Bienen, PhD, of The Curry Rockefeller Group, LLC, Tarrytown, New York, for medical editorial assistance with this report. Editorial support was funded by Acorda Therapeutics, Inc. These studies were conducted at XenoTech, LLC (Lenexa, Kansas).
References
- Ampyra® [dalfampridine] extended release tablets prescribing information. Ardsley, NY: Acorda Therapeutics, Inc., January 2013
- Goodman AD, Brown TR, Krupp L, et al. Sustained release of oral fampridine in multiple sclerosis: a randomised, double-blind, controlled trial. Lancet 2009;373:732-8
- Goodman AD, Brown TR, Edwards KR, et al. A phase 3 trial of extended release oral dalfampridine in mulitple sclerosis. Ann Neurol 2010;68:494-502
- Dunn J, Blight A. Dalfampridine: a brief review of its mechanism of action and efficacy as a treatment to improve walking in patients with multiple sclerosis. Curr Med Res Opin 2011;27:1415-23
- Lundh H, Thesleff S. The mode of action of 4-aminopyridine and guanidine on transmitter release from motor nerve terminals. Eur J Pharmacol 1977;42:411-2
- Kirsch GE, Drewe JA. Gating-dependent mechanism of 4-aminopyridine block in two related potassium channels. J Gen Physiol 1993;102:797-816
- Mahmood I, Green MD. Drug interaction studies of therapeutic proteins or monoclonal antibodies. J Clin Pharmacol 2007;47:1540-54
- Schapiro R, Brown T, Goodman A; On Behalf of the MS-F202 M-F, and MS-F204 Study Groups. Evaluation of orally administered prolonged-release fampridine tablets for walking improvement in patients with multiple sclerosis treated with disease-modifying therapies. [abstract]. Mult Scler 2010;16(Suppl):S174
- Vollmer T, Henney HR. Pharmacokinetics and tolerability of single escalating doses of fampridine sustained-release tablets in patients with multiple sclerosis: a phase I-II, open-label trial. Clin Ther 2009;31:2206-14
- Vollmer T, Blight AR, Henney HR. Steady-state pharmacokinetics and tolerability of orally administered fampridine sustained release 10-mg tablets in patients with multiple sclerosis: a 2-week, open-label, follow-up study. Clin Ther 2009;31:2215-23
- Blight AR, Henney HR. Pharmacokinetics of 14C-radioactivity after oral intake of a single dose of 14C-labeled fampridine (4-aminopyridine) in healthy volunteers. Clin Ther 2009;31:328-35
- Smith W, Swan S, Marbury T, et al. Single-dose pharmacokinetics of sustained-release fampridine (Fampridine-SR) in healthy volunteers and adults with renal impairment. J Clin Pharmacol 2010;50:151-9
- Henney HR 3rd, Blight A, Faust B. Effect of food on the single-dose pharmacokinetics and tolerability of dalfampridine extended-release tablets in healthy volunteers. Am J Health Syst Pharm 2011;68:2148-54
- United States Food and Drug Administration. Guidance for Industry. Drug Interaction Studies — Study design, data analysis, and implications for dosing and labeling. Draft Guidance. US Department of Health and Human Services. Rockville, MD: Center for Drug Evaluation and Research; September 2006
- Ogilvie BW, Zhang D, Li W, et al. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: implications for drug-drug interactions. Drug Metab Dispos 2006;34:191-7
- Ogilvie BW, Usuki E, Yerino P, et al. In vitro approaches for studying the inhibition of drug-metabolizing enzymes and identifying the drug-metabolizing enzymes responsible for the metabolism of drugs (reaction phenotyping) with emphasis on cytochrome P450. In: Rodrigues AD, ed. Drug-drug interactions drugs and the pharmaceutical sciences. 2nd edn. New York: Informa Healthcare, 2008. p 231-358
- Quistorff B, Dich J, Grunnet N. Preparation of isolated rat liver hepatocytes. In: Pollard JW, Walker JM, eds. Methods in molecular biology. Volume 5. Animal cell culture. Clifton, NJ: Humana Press. 1989. p 151-60
- Madan A, Graham RA, Carroll KM, et al. Effects of prototypical microsomal enzyme inducers on cytochrome P450 expression in cultured human hepatocytes. Drug Metab Dispos 2003;31:421-31
- Sonderfan AJ, Parkinson A. Inhibition of steroid 5 alpha-reductase and its effects on testosterone hydroxylation by rat liver microsomal cytochrome P-450. Arch Biochem Biophys 1988;265:208-18
- Evenhuis J, Agoston S, Salt PJ, et al. Pharmacokinetics of 4-aminopyridine in human volunteers. A preliminary study using a new GLC method for its estimation. Br J Anaesth 1981;53:567-70
- Uges DR, Sohn YJ, Greijdanus B, et al. 4-Aminopyridine kinetics. Clin Pharmacol Ther 1982;31:587-93
- Caggiano A, Blight A. Identification of metabolites of dalfampridine (4-Aminopyridine) in human subjects and reaction phenotyping of relevant cytochrome P450 pathways. J Drug Assessment 2013