
Abstract
Context: Breast cancer accounts for 23% of all newly occurring cancers in women worldwide and represents 13.7% of all cancer deaths. Available chemotherapeutic agents are limited largely due to the low accumulation of chemotherapeutics at the tumors relative to their accumulation at normal (healthy) organs due to developed multidrug resistance (MDR) which translates into increased toxicities. Objectives: To enhance the anticancer potency of docetaxel (DTX), by encapsulating it inside the nanosize lipid particles (folate-conjugated PEG-solid fat nanoemulsions) stabilized by soya phosphatidylcholine (PC) for targeting folate receptors in breast cancer. Materials and methods: Tristearin and soya PC-based solid fat nanoemulsions were prepared by cast film technique followed by sonication and modified by coating them with folate receptor-specific ligand (folate-PEG-cholesterol). The surface modified solid fat nanoemulsions and their plain counterparts were characterized for size, shape, lamellarity, zeta potential and entrapment efficiency using scanning electron microscopy, zetasizer and minicolumn centrifugation. The in vitro release profile (dialysis bag technique) and in vitro cytotoxicity (MTT assay) of the developed formulations were evaluated. Results: The TEM photograph showed homogenous and spherical nature of the particles. The IC50 values of docetaxel solution, plain solid fat nanoemulsions, and folate-conjugated PEG-solid fat nanoemulsions were found to be 14.2, 64.9, and 28.8 μM/ml, respectively. Strong fluorescence was observed in the HeLa cells treated with folate-conjugated PEG-solid fat nanoemulsions which showed higher cellular uptake of ligand-appended solid fat nanoemulsions than plain solid fat nanoemulsions. Discussion and conclusion: The results indicated that folate was effective in promoting the internalization of solid fat nanoemulsions encapsulating DTX to the folate receptor-positive tumor cells. This opens the new possibility for non-immunogenic, site-specific delivery of bioactive(s).
Introduction
Breast cancer is a major health problem in women and the fifth most common cause of cancer death. Worldwide, it is the most common type of non-skin cancer, which comprises 10.4% of all cancer incidences among women and caused 519,000 deaths worldwide. More than half of the cases are in developed countries. In United States, 75% of new cases of breast cancer are in women aged 50 years or older. Within the last 5 years, there are 4.4 million women alive having breast cancer. It has been estimated that more than 1.5% of the US female population is suffering from breast cancer and it is about 100 times more widespread in women than in men (CitationFumoleau et al. 1997).
Although excellent chemotherapeutic agents against breast cancer are available, chemotherapy is third to surgery and radiation, largely due to two main reasons. First, the low accumulation of chemotherapeutics at the tumors relative to their accumulation at normal (healthy) organs which translates into increased toxicities. And second, the low drug bioavailability within the cancer cells that constitutes the tumors owing to the inability of drugs to reach their molecular sites of action within the malignant cells due to developed multidrug resistance (MDR). Therefore, novel approaches for the aggressive treatment of breast carcinoma are urgently needed to address these issues and to increase the survival rates.
Solid fat nanoemulsions have been extensively investigated as carriers for the treatment of visceral leishmaniasis, using amphotericin B (CitationGupta et al. 2007). Solid fat nanoemulsions are pharmaceutical compositions comprising nanoemulsions of particles comprising a lipid core composed of lipid which is in a solid or liquid crystalline phase at at least 25°C, stabilized by at least one phospholipid envelope. Solid fat nanoemulsion formulations serve as circulating reservoir of cytotoxic agents and thus prevent passive diffusion of the encapsulated moiety into healthy tissues/cells, and as a result, circulation time increases and undesired side effects associated with them decreases. Solid fat nanoemulsions can increase drug delivery to specific target such as tumors through a passive targeting mechanism which takes advantage of the enhanced vascular permeability and impaired lymphatic drainage in growing tumors via the enhanced permeation and retention effect. However, solid fat nanoemulsions have been found to be associated with rapid opsonization and are taken up by the reticuloendothelial system cells located mainly in the liver and spleen due to their colloidal size. This problem can be resolved by incorporating lipid-grafted polyethylene glycol (PEG) into the nanoemulsion membrane. The modification of the solid fat nanoemulsions surface by the flexible polyethylene glycol makes them stealth drug carriers devoid of opsonization and long circulatory in nature (CitationMaeda et al. 2000, CitationAggarwal et al. 2011).
Active targeting strategies using a ligand coupled to the surface of solid fat nanoemulsions, which could be taken up by cancer cells via a receptor-mediated pathway, may improve the performance of DTX-loaded solid fat nanoemulsions. Tumor cells commonly overexpress receptors like epidermal growth factor receptor, folate receptor (FR), transferrin receptor, fucose receptor, estrogen receptor, progesterone receptor, thrombin receptor, vasoactive intestinal peptide receptor, etc. (CitationRojo et al. 2008). Targeting the folate receptor has shown considerable promise in mediating the uptake of a variety of drugs when folic acid is conjugated to the drug or delivery vehicle (CitationYuan et al. 2010). The folic acid receptors are overexpressed by many types of tumor cells, including ovarian, endometrial, colorectal, breast, lung, renal, neuroendocrine carcinomas, and brain metastases (CitationCaliceti et al. 2003).
Folic acid is a vitamin required for one-carbon transfer reactions in several metabolic pathways. Because folic acid is essential for the biosynthesis of nucleotide bases, the vitamin is consumed in elevated quantities by proliferating cells (CitationLu and Low 2002). Differential expression of the folate receptor has been exploited to target liposomes to tumors. Tumor cells that vastly overexpress the folate receptor showed significant uptake of folate-targeted anti-cancer drug, while normal cells that do not express the folate receptor showed minimal uptake of the drug (CitationYuan et al. 2010).
While overexpression of FR on many cancer cells obviously identifies the receptor as a potential target for a variety of ligand- and antibody-directed cancer therapeutics, FR may be further qualified as a tumor-specific target, since it generally becomes accessible to intravenous drugs only after malignant transformation. That is, because FR is selectively expressed on the apical membrane surface of certain epithelial cells (i.e., the membrane surface facing a body cavity), it is inaccessible to blood-born reagents and therefore protected from FR-directed therapeutics delivered in the plasma. However, upon epithelial cell transformation, cell polarity is lost and FR becomes accessible to targeted drugs in circulation. Probably because of this dual mechanism for tumor specificity, the receptor's natural ligand, folic acid, has become a popular molecule for targeting drugs attached to cancer cells. The attractiveness of folate has been further enhanced by its high binding affinity, low immunogenicity, ease of modification, small size, stability during storage, compatibility with a variety of organic and aqueous solvents, low cost, and ready availability. In virtue of its ability to be taken up by folate receptor overexpressing tumor cells, folic acid has been widely investigated as targeting molecule for active anticancer drug delivery. Lu and Low were the first to report the synthesis of folate-conjugated liposomes. Proper synthesis procedures have been pointed out to link folic acid to drug carriers to develop targeted drug delivery systems (CitationCaliceti et al. 2003, CitationLu and Low 2002). When coupled to the terminal end of PEG chains of liposomes, folic acid and other targeting molecules have the flexibility to interact with their corresponding cell surface receptor to mediate liposomal uptake via endocytosis (CitationSaul et al. 2003).
DTX binds to the subunit of tubulin, which inhibits depolymerization and induces polymerization of the tubulin into stable microtubules. Therefore, these modifications are lethal resulting in an accumulation of cells in the G2/M phase, disrupting mitosis and cell replication (CitationGuo et al. 2000).
DTX has been found to be effective against breast, ovarian, lung, and head and neck cancers. To overcome the poor solubility of DTX and to improve its efficacy, novel formulations of DTX have been developed. DTX-loaded liposomes have been prepared and showed a better antitumor activity and a longer circulation time in the body. A pegylated liposomal formulation of DTX has also been developed with the purpose of improving DTX solubility without any need to use Tween 80 and of enhancing stability in the blood circulation. Although these formulations have some advantages, there are some problems that need to be solved to meet the requirements for clinical use and industrial production. Cholesterol is frequently included in liposomes and solid fat nanoemulsions to control the release of the entrapped drugs or to enhance the stability of the formulations (CitationYuan et al. 2010). In this study, a folate-PEG-cholesterol was synthesized, and solid fat nanoemulsions containing this reagent were evaluated.
Materials and methods
Materials
DTX was a kind gift sample from Sun Pharma, Vadodara, India. Folic acid, bis-amine-PEG (Mw- 3,350), cholesteryl chloroformate, N-hydroxysuccinimide (NHS), dimethylaminopropyl ethyl carbodiimide (EDC), soya PC, sephadex G-50, dialysis membrane (molecular weight cutoff, 12 kDa) and cholesterol were procured from Sigma Aldrich, Mumbai (India). Tristearin and 3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazoliumbromide (MTT) was obtained from HiMedia Laboratories Pvt. Ltd. Mumbai (India). Chloroform and all other chemicals were of pure analytical grade and used as procured.
Preparation of folate anchored solid fat nanoemulsions
Synthesis of folate- PEG-cholesterol
Conjugation of folate-PEG-cholesterol was done in two steps with slight modifications in the process described (CitationGuo et al. 2000). First, folic acid was conjugated with bis-amine PEG, and in second step, folate-PEG-NH2 was further conjugated to cholesteryl chloroformate ().
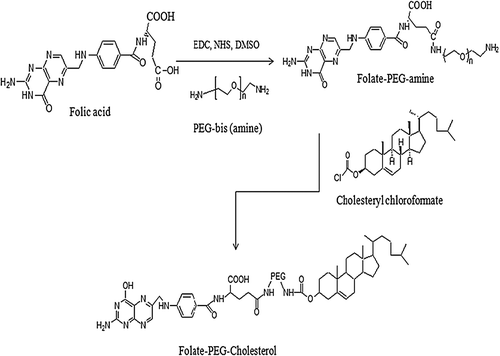
Preparation of folate-PEG-NH2
For the synthesis of the folate-PEG-amine, folic acid was dissolved in dimethylsulfoxide(DMSO, 25 mg/ml) along with 1.1 M excess of EDC and 1M eq. of NHS, and the reaction mixture was stirred for 20 min in dark.
Then, 1M eq. of bis-amine PEG was added to the reaction mixture and stirred for 24 h in dark at room temperature. The progress of reaction was monitored using pre-coated UV-active thin-layer chromatography plates. The folate-PEG-amine was extracted in the mixture of chloroform/water (1:1). Both water and chloroform layers were repeatedly extracted with fresh chloroform and water thrice. Chloroform layer was then washed with anhydrous sodium sulfate to remove traces of water present in layer. Chloroform layer was then concentrated under vacuum and allowed to precipitate with the addition of non-solvent, diethyl ether. The conjugate was further characterized by 1H-NMR using DMSO as a solvent on Bruker Avance II 400 MHz NMR spectrometer (CitationGuo et al. 2000).
Preparation of folate-PEG-cholesterol
To synthesize folate-PEG-cholesterol, a 1.1 molar excess of cholesteryl chloroformate was added to folate-PEG-amine in chloroform. The reaction was carried out overnight at room temperature. Progression of the reaction was monitored by the disappearance of free amino group, which was followed by ninhydrin assay. The product, folate-PEG-cholesterol, was dried under vacuum, washed twice with ether to remove any residual cholesteryl chloroformate, and stored at − 20°C. The purity of the product was analyzed by silica gel thin-layer chromatography using a solvent system composed of chloroform/methanol (8:2) with trace amount of ammonia. The concentration of folate-PEG-cholesterol was determined by measuring the folate content in the product using UV extinction at 363 nm (CitationGuo et al. 2000). Final product was further characterized by 1H-NMR using DMSO as a solvent on Bruker Avance II 400 MHz NMR spectrometer.
Preparation of solid fat nanoemulsions
Solid fat nanoemulsions containing DTX were prepared by already established method (CitationAmselem et al. 1994, CitationAmselem and Friedman 1997, CitationPal et al. 2012, CitationGupta and Vyas 2007), with slight modifications as per our laboratory setup. In a beaker PC, tristearin, DTX, and cholesterol were co-dissolved in chloroform and transferred to a 250-ml round-bottomed flask, and the organic solvent was evaporated until complete dryness under reduced pressure using a rotary flash evaporator to form a thin lipid film on the walls of the round-bottom flask. The dried film was hydrated with phosphate-buffered saline (PBS) of pH 7.4 (4 ml), and homogenized by ultrasonication at 50% amplitude to obtain solid fat nanoemulsions of nanometric size range. The free unentrapped drug was removed by passing the dispersion through a sephadex G-50 column (CitationFry et al. 1978, CitationNew RRC 1990, CitationVyas et al. 2006).
Optimization of solid fat nanoemulsions
Solid fat nanoemulsions were optimized for DTX to solid lipid ratio, PC to tristearin ratio, and sonication time to determine their influence on the particle size and entrapment efficiency. For optimization of DTX to lipid ratio, PC to tristearin ratio (1:1 w/w), cholesterol (4% w/w of PC), and sonication time (4 min) were kept constant while DTX content was varied at different weight percent ratio levels (i.e., 1, 2, 3, 4, 5, 6% weight of the solid lipid) in different formulations for determining optimum DTX content (CitationVyas et al. 2006, CitationVyas et al. 2005). Average particle size (Delsa NanoC, Beckman Coulter Pvt. Ltd.) and percent drug entrapment of different formulations was determined (CitationVyas et al. 2000). Solid fat nanoemulsions with optimum DTX to lipid ratio were optimized for optimum sonication time in terms of average particle size. PC to tristearin ratio (1:1 w/w), cholesterol (4% w/w of PC) and DTX to lipid ratio (optimized) were kept constant, while sonication time was varied (i.e., 0, 2, 4, 6, and 8 min) for different formulations. Solid fat nanoemulsions were evaluated for average particle size and polydispersity index. Solid fat nanoemulsions with optimum DTX to lipid ratio and sonication time were optimized for optimum PC to tristearin ratio in terms of percent drug entrapment. The solid fat nanoemulsions with fixed cholesterol (4% w/w of PC) and different PC to tristearin ratios (0.6:1, 0.8:1, 1:1, 1.2:1 and 1.4:1 w/w ratios) were prepared. DTX content and sonication time, however, were kept constant at its optimum level. Solid fat nanoemulsions were evaluated for percent drug entrapment (CitationVyas et al. 2000, CitationVyas et al. 2005). Percent drug entrapments were plotted against PC to tristearin ratio, from which optimum PC to tristearin ratio was determined.
Optimized formulation was further used to prepare ligand-appended solid fat nanoemulsions which were prepared by the hydration of a dried, thin layer of lipid (consisting of a PC, cholesterol, tristearin and folate- PEG-cholesterol, and DTX).
Characterization of solid fat nanoemulsions
Particle size and morphology
The formulations were characterized for their shape and surface morphology using scanning electron microscopy (SEM) (Jeol, Japan). A drop of the sample was placed on to a grid to leave a thin film on the grid. The grid was allowed to dry at ambient temperature, and samples were viewed under SEM and photographs were taken at suitable magnification. Formulations were also visualized under transmission electron microscopy (TEM) with an accelerating voltage of 100 Kv (CitationNew RRC 1990). A drop of the sample was placed on to a carbon-coated copper grid to leave a thin film on the grid. The film was negatively stained with 1% phosphotungstic acid solution and dried. A drop of the staining solutions was added on to the film, and the excess of the solution was drained off. The grid was allowed to dry at ambient temperature and samples were viewed under TEM (Hitachi, Japan) and photographs were taken at suitable magnification. The mean particle size was measured using zetasizer (Bechman coulter, Delsa NanoC).
Entrapment efficiency
Sephadex G-50 (1 g) was allowed to swell in 10 ml of 0.9% sodium chloride solution for 5 h at room temperature with occasional shaking. The gel so formed was stored at 4°C. The hydrated gel was filled to the top in the barrel of 1 ml of disposable syringe plugged with whatman filter pad. The barrel was then placed in centrifuge tubes. The tubes were centrifuged at 3000 rpm for 3 min to remove the excess saline solution. Percent drug entrapment was determined and expressed as the ratio of experimentally measured amount of drug in dispersion and initial amount used for entrapment; 0.2 ml of formulation (undiluted) was applied drop wise on the top of the gel bed in the center. Columns were centrifuged at 3000 rpm for 3 min to remove unentrapped drug. Elute was removed and 0.2 ml saline was applied to each column, and centrifuged as previously mentioned. Amount of drug entrapped in the solid fat nanoemulsions was then determined by disrupting the solid fat nanoemulsions by adding 1.0 ml of 0.1% v/v Triton X-100 and assayed for drug content using UV spectrophotometer at 228 nm (CitationNew RRC 1990, CitationPal et al. 2012).
The percentage drug entrapment was measured as:
In-vitro drug release study
The in-vitro drug release profile of entrapped drug from plain and ligand appended solid fat nanoemulsions was studied in PBS/DMSO (95:5) pH (7.4) using dialysis membrane (molecular weight cutoff, 12 kDa; Sigma Chemical Co., USA) (CitationVyas et al. 2007). Formulations were first centrifuged in a mini-column laboratory centrifuge to remove unentrapped drug. Pure solid fat nanoemulsions dispersion (2 ml), free of any unentrapped drug, were taken into a dialysis bag (donor compartment) and placed in a beaker containing 100 ml of PBS/DMSO (95:5) (receptor compartment). The temperature of beaker was maintained at 37 ± 1°C, under stirring at 100 rpm throughout the study. Samples (2 ml) were withdrawn at predetermined time interval and replaced with the same volume of fresh dialyzing medium PBS/DMSO (95:5) while maintaining strict sink conditions, and the withdrawn samples were assayed for drug content by measuring absorbance at 228 nm against the blank using UV-1700 spectrophotometer.
Cytotoxicity study (MTT assay)
HeLa cells were obtained from NCCS, Pune. Cell viability was tested using MTT assay based on the cleavage of yellow tetrazolium salt MTT by metabolically active cells to form an orange formazan dye which was quantified using ELISA (Enzyme linked immunosorbent assay) (CitationMosmann 1983). Cells were seeded in 96-well microtitre plates (2 × 104 cells/200 μl growth medium/well) followed by overnight incubation. Supernatants from the wells were aspirated out and were replaced with medium containing serial dilutions of DTX formulation: folate-conjugated PEG-solid fat nanoemulsions, plain solid fat nanoemulsions and free DTX, respectively. Experiment was done in triplicate. After 24 h, supernatants were aspirated out and the cell monolayers in the wells were washed with 200 μl PBS (0.1 M, pH 7.4). Subsequently, MTT reagent (150 μl, 0.8 mg/ml) was added in each well and incubated for 5 h. DMSO (100 μl) was added in each well after aspirating out the supernatant and incubated at 37°C for 1 h. Absorbance at two wavelengths (570 nm for soluble dye and 630 nm for cells) was recorded using ELISA. Concentrations of samples showing 50% reduction in cell viability (i.e., IC50 values) were then calculated.
Cell uptake study
The solid fat nanoemulsions encapsulating fluorescent marker (fluorescein isothiocyanate [FITC]) were prepared. Folate-positive HeLa cells were seeded on glass coverslips (18 × 18 mm) placed into 35-mm tissue culture plates at a density of 2 × 105 cells (in 2-ml growth medium). After overnight growth, supernatants from the culture plates were aspirated out and fresh aliquots of growth medium containing suspension of FITC-loaded plain solid fat nanoemulsions and folate-conjugated PEG-solid fat nanoemulsions were added. Upon incubation for 6 h, cells were washed with PBS and fixed in 4% formaldehyde for 20 min and then washed twice with PBS and observed under inverted microscope (Olympus, CKX41, USA) and photomicrographs were taken.
Statistical analysis
All the experiments were carried out thrice, independently. The data obtained were expressed in terms of ‘mean ± SD’ values. Wherever appropriate, the data were also subjected to unpaired two tailed Student's t-test. A value of p < 0.05 was considered as significant.
Results
Preparation of folate-PEG-cholesterol conjugate
Folate-PEG-cholesterol was synthesized in two step reaction. First, conjugation of folic acid with bis-amine PEG was conducted using NHS and EDC as coupling agents. Product was extracted in chloroform layer and precipitated by the addition of diethyl ether. When analyzed using silica gel thin layer chromatography, the folate-PEG-amine was appeared as a single spot with an Rf of 0.82. Further confirmation was done using 1H-NMR spectroscopy of folate-PEG-amine (). In second step, folate-PEG-amine was further conjugated to cholesteryl chloroformate as described earlier. shows the 1H-NMR of the final conjugate, that is, folate-PEG-cholesterol. The interpretation revealed the presence of folic acid by the signals in 6.61–8.63 regions. Incorporation of cholesterol was confirmed from the integration value for the protons between 0.82 and 4.48 regions, which included the signals for the protons of PEG. In comparison with 1H-NMR for folate-PEG-amine and the final NMR, it can be concluded that the conjugation of cholesterol to folate-PEG-amine has been achieved.
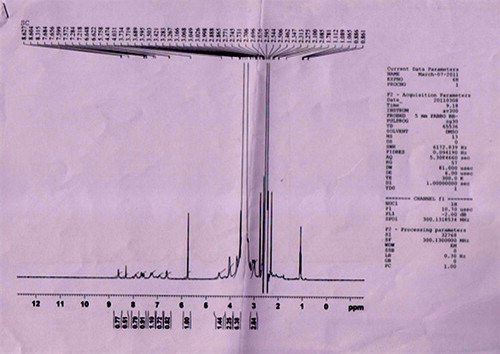
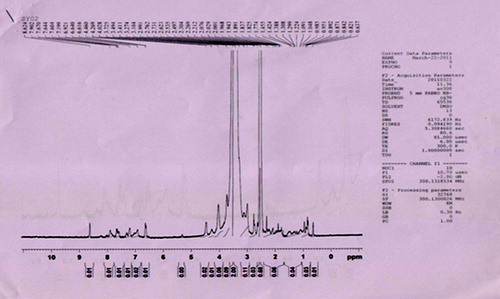
Preparation and optimization of solid fat nanoemulsions
Solid fat nanoemulsions were prepared as per the method described earlier and optimized for drug to lipid ratio, PC to tristearin ratio, and sonication time.
Optimization of drug to lipid ratio
In order to optimize the DTX to lipid ratio, solid fat nanoemulsions were prepared and evaluated for particle size and entrapment efficiency as shown in .
Table I. Optimization of drug to lipid ratio.
Optimization of sonication time
In order to optimize the sonication time, solid fat nanoemulsions formulations were prepared and evaluated for particle size as shown in .
Table II. Optimization of sonication time.
Optimization of PC to tristearin ratio
In order to optimize the PC to tristearin ratio, solid fat nanoemulsions were prepared and evaluated for percent drug entrapment as shown in .
Table III. Optimization of PC to tristearin ratio.
The optimum ratios of PC/tristearin/cholesterol/folate-PEG-cholesterol were found to be 24:20:0.96:1 (mg). The SEM and TEM photographs showed homogenous and spherical nature of the plain and ligand-anchored solid fat nanoemulsions (). The mean diameter of folate-conjugated PEG-solid fat nanoemulsions and plain solid fat nanoemulsions were 230 ± 2.3 nm and 182.7 ± 3.63 nm with narrow size distribution as indicated by polydispersity index of 0.287 ± 0.034 and 0.234 ± 0.14, and entrapment efficiency was found to be 81.42 ± 2.52 and 83.26 ± 3.12%, respectively ().
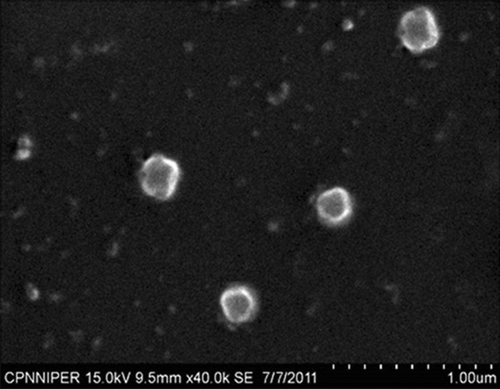
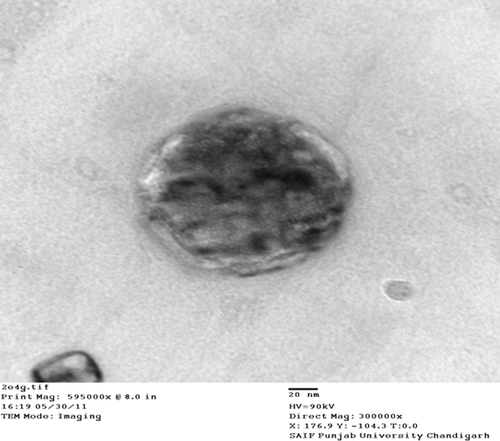
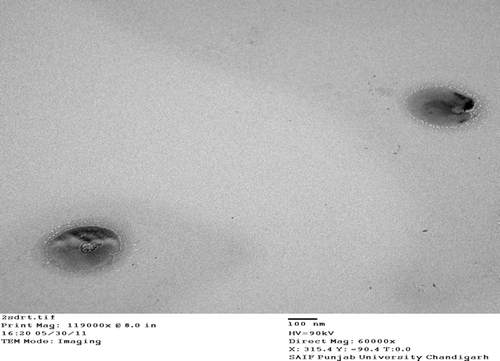
Table IV. Various characterization parameters for optimized formulation of DTX-loaded solid fat nanoemulsions (mean ± SD, n = 3).
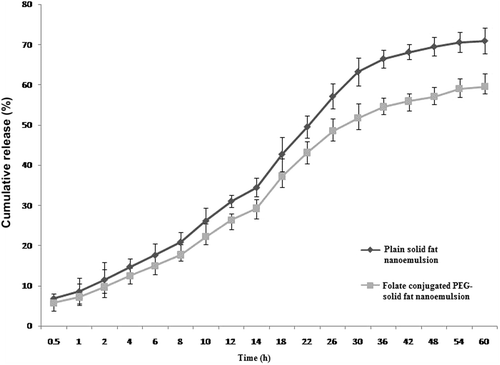
The in vitro drug release profile of the drug from both formulations was determined in PBS/DMSO (95:5) of pH 7.4 for a period of 60 h. The cumulative percent drug released from the folate-conjugated PEG-solid fat nanoemulsions and plain solid fat nanoemulsions was found to be 52 ± 1.8 and 70.92 ± 0.23%, respectively (). Various parameters of DTX-loaded solid fat nanoemulsions are presented in .
Cytotoxicity study
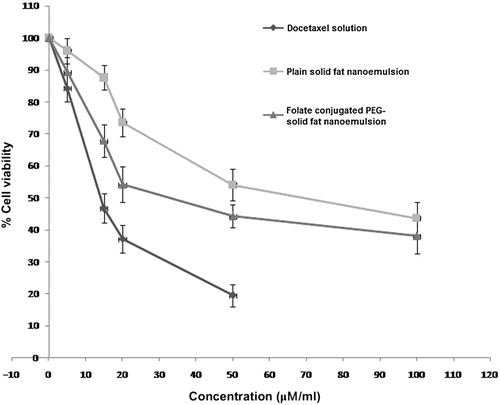
Cytotoxicity of folate-conjugated PEG-solid fat nanoemulsions, plain solid fat nanoemulsions, and free DTX was determined in HeLa cells using MTT assay. shows the concentrations of DTX either in solution or in various solid fat nanoemulsions that caused 50% inhibition of cell growth after a 24-h exposure. Cell viability in HeLa cells revealed an effective suppression of cell proliferation by the treatment of DTX-loaded solid fat nanoemulsions. No cytotoxicity of the plain solid fat nanoemulsions or a DTX solution was observed in cell-line study. The concentrations of DTX required to achieve 50% growth inhibition in cell line were much lower in solution than in the solid fat nanoemulsions. The IC50 values of DTX solution, plain solid fat nanoemulsions, and folate-conjugated PEG-solid fat nanoemulsions were found to be 14.2, 64.9, and 28.8 μM/ml, respectively. The cytotoxicity of folate-conjugated PEG-solid fat nanoemulsions on HeLa cells was sufficiently high in comparison with that of plain solid fat nanoemulsions, and this was most probably attributed to the faster uptake of folate-conjugated PEG-solid fat nanoemulsions.
Cell uptake study
Strong fluorescence was observed in the HeLa cells treated with folate-conjugated PEG-solid fat nanoemulsions, whereas the fluorescence of the plain solid fat nanoemulsions was noticeably much weaker () which showed higher cellular uptake of ligand-appended solid fat nanoemulsions than plain solid fat nanoemulsions.
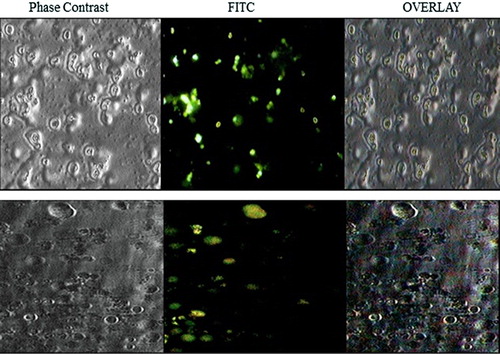
Discussion
As folate receptors are overexpressed in various cancers, and folic acid (folate) is a high-affinity ligand for the FRs that retains high FR affinity upon derivatization via one of its carboxyl groups. Due to its small size and ready availability, folate has become one of the most investigated targeting ligands for tumor-specific drug delivery. Folate has been incorporated into liposomes via conjugation to lipophilic anchors. Folate-conjugated liposomes have been evaluated for targeted delivery of a broad range of therapeutic agents. Therefore, it may act as potential target for folate-anchored drug delivery (CitationXiang et al. 2008). DTX is one of the most widely used broad spectrum antineoplastic agents, and it has been used clinically against a wide variety of cancers. The main toxic effects of DTX as expected with an antimitotic agent that alters microtubule depolymerization were most evident in tissues with a high cell turnover (hematopoietic, lymphatic, testicular, and hair cells) or in those where microtubules play an important functional role (e.g., neuronal). Various issues such as neutropenia, anemia and thrombocytopenia, drug resistance and non-specificity toward the desired site are critical to its therapeutic success and safety of the drug, which remain to be improved. The lower toxicity of DTX-loaded liposomes in comparison with that of free DTX is advantageous (CitationFumoleau et al. 1997). Surface modification with hydrophilic polymer enhances blood circulation time of solid fat nanoemulsions by preventing reticulo-endothelial system uptake, but the desired success is still awaited. Site-specific targeting of colloidal carriers to the expressed receptors over the tumor surface is the strategy that can be utilized to increase drug bioavailability and biodistribution (CitationAggarwal et al. 2011). Thus, intracellular delivery of DTX into tumor cells will be essential to its antitumor activity.
This study reports development and characterization of folate-targeted stealth nanoemulsion formulation of DTX, which possesses specificity toward folate-responding cells. The incorporation of PEG can extend the plasma circulation half-life of the nanoemulsion formulation by shielding the particle surface against opsonization and reducing the uptake by the reticuloendothelial system. Data in this report showed that stable solid fat nanoemulsion with narrow size distribution and high drug incorporation can be prepared using this composition. Folate receptor acts as a tumor marker that is consistently overexpressed in breast/ovarian carcinomas.
One of the important constituent of solid fat nanoemulsions, folate-PEG-cholesterol, was prepared by the previously reported method using simple conjugation chemistry with a slight modification. Solid fat nanoemulsion formulations, based on PC and tristearin as the constitutive lipids, were prepared. The drug DTX was incorporated into the solid fat nanoemulsions using cast film method. Solid fat nanoemulsions were optimized for various parameters. These include the weight ratio of DTX to total lipid, sonication time, and ratio of PC to tristearin.
At higher concentration, (6% weight of the total lipid) of DTX, lower entrapment efficiency and lower size of the particles were found (65.70 ± 4.5% and 275.2 ± 2.0 nm, respectively). When the concentration of DTX was used at 4% weight of total lipid, average particle size measured was 355.3 ± 2.4 nm and entrapment efficiency was recorded to be 75.3 ± 5.2% (). Below this concentration, (i.e., 2% and 1% weight of DTX to total lipid) percent drug entrapment as well as particle size were recorded to be increased (82.3 ± 1.5 and 86.24 ± 3.2% and 506.5 ± 3.8 and 728.3 ± 4.3 nm, respectively). But formulation having DTX concentration 5% weight of total lipid (formulation E5) was considered to be optimum having acceptable particle size and high drug entrapment (CitationGupta et al. 2007). Formulation with optimum DTX to lipid ratio was subjected to sonication for different time periods to optimize the sonication time (). It was seen that as the sonication time was increased from 0 to 8 min, average particle size was recorded to be decreased. Optimum sonication time was recorded to be 6 min, which gave particle size of 198.9 ± 0.75 nm. On further increasing the sonication time (i.e., at 8 min) beyond the optimum limit, constant or a little increase in the particle size was recorded. Formulation having optimized DTX to lipid ratio and sonication time was subjected to optimization of PC to tristearin ratio. As the PC: tristearin w/w ratio was increased from 0.6:1 to 1.2:1, percent drug entrapment was also recorded to be increased (). This may be attributed to the formation of more bilayers upon increasing the concentration of PC and intercalation of DTX in the bilayers also apart of internal solid lipid core (CitationPal et al. 2012). Optimum PC: tristearin w/w ratio was found to be 1.2:1 which could entrap maximum amount of drug (83.26 ± 3.12%). On further increasing the PC content (i.e., 1.4:1 w/w PC: tristearin) beyond this optimum limit, percent drug entrapment was recorded to be greatly reduced (i.e., 64.12 ± 1.5%). The higher PC content might have formed much more unstable particles with greater permeability which subsequently resulted to the fast leakage of the entrapped drug therein (CitationNew RRC 1990; CitationVyas et al. 2006). Parameters for in vitro characterization of different nanoemulsion formulations include size, shape, size distribution, entrapment efficiency, and in vitro drug release profile. Mean size of a colloidal drug carrier is an important requisite that may influence the in vivo performance of delivery system (CitationAggarwal et al. 2011). In order to obtain solid fat nanoemulsion formulation with a reduced mean size, the sonication with probe sonicator was carried out. Sonication followed by extrusion reduced the size of solid fat nanoemulsions into nanorange and also allowed the formation of solid fat nanoemulsion with narrow size distribution. TEM of formulation suggested that the particles were spherical in shape. The polydispersity index of the formulations was found to be < 0.3, which showed uniformity of preparations. Entrapment efficiency of DTX in both the formulations was more than 80%. In vitro drug release study was performed in PBS/DMSO (95:5) pH 7.4 for a period of 60 h. A slow but consistent release was obtained which might be due to the release of drug from PC bilayers surrounding the lipid core and later on prolonged release could be attributed to the release of drug from the solid lipid core. The in vitro cytotoxicity of folate-conjugated PEG-solid fat nanoemulsions was compared with the cytotoxicity of plain solid fat nanoemulsions and free DTX. The IC50 value of targeted folate-conjugated PEG-solid fat nanoemulsions was significantly (P < 0.05) lower than that of plain solid fat nanoemulsions in folate receptor-positive HeLa cells. These results indicated that folate was effective in promoting the internalization of solid fat nanoemulsions encapsulating DTX to the folate receptor-positive target tumor cells. Folate-conjugated PEG-solid fat nanoemulsions provided the most efficient killing of cancer cells compared to plain solid fat nanoemulsions via receptor-mediated targeting. The cellular uptake of folate-conjugated PEG-solid fat nanoemulsions was higher than that of plain solid fat nanoemulsions.
Conclusions
FR-targeted formulation of antineoplastic agents may be potentially useful in the treatment of FR-positive tumors such as breast cancer. This opens the new possibility for non-immunogenic, site-specific delivery of bioactive(s). Further studies are warranted to investigate the potential therapeutic advantage of the FR-targeted nanoemulsion formulation and possible mechanism of in vivo tumor targeting
Acknowledgements
The authors acknowledge Chairman, ISF College of Pharmacy, Moga, for providing the necessary facilities required to carry out this research work.
Declaration of interest
The authors report no declaration of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Aggarwal S, Yadav S, Gupta S. 2011. EGFR targeted PLGA nanoparticles using gemcitabine for treatment of pancreatic cancer. J Biomed Nanotechnol. 7:137–138.
- Amselem S, Yogev A, Zawoznik E, Friedman D. 1994. Emulsomes, a novel drug delivery technology. Proc Int Symp Control Rel Bioact Mater. 21:1368–1369.
- Amselem S, Friedman D. 1997. Solid fat nanoemulsions. United States Patent US 5662932.
- Caliceti P, Salmaso S, Semenzato A, Bersani S, Matricardi P, Rossi F. 2003. Synthesis and physicochemical characterization of folate- cyclodextrin bioconjugate for active drug delivery. Bioconjug Chem. 14:899–908.
- Fumoleau P, Seidman AD, Trudeau ME, Chevallier B, Ten BHWW. 1997. Docetaxel: a new active agent in the therapy of metastatic breast cancer. Expert Opin Investig Drugs. 6:1853–1865.
- Fry DW, White JC, Goldman ID. 1978. Rapid separation of low molecular weight solutes from liposomes without dilution. Anal Biochem. 90:809–815.
- Gupta S, Dube A, Vyas SP. 2007. Antileishmanial efficacy of amphotericin B bearing emulsomes against experimental visceral leishmaniasis. J Drug Target. 15:437–444.
- Gupta S, Vyas SP. 2007. Development and characterization of amphotericin B bearing emulsomes for passive and active macrophage targeting. J Drug Target. 15:206–217.
- Guo W, Lee T, Sudimack J, Lee RJ. 2000. Receptor-specific delivery of liposomes via folate-peg-chol. J Liposome Res. 10:179–195.
- Lu Y, Low PS. 2002. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv Drug Deliv Rev. 54:675–693.
- Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. 2000. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Contr Release. 65:271–284.
- Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 65:55–63.
- New RRC. 1990. Introduction and preparation of liposomes. In: New RRC, Ed. Liposomes: A Practical Approach. Oxford: IRL Press, pp. 1–104.
- Pal A, Gupta S, Jaiswal A, Dube A, Vyas SP. 2012. Development and evaluation of tripalmitin emulsomes for the treatment of experimental visceral leishmaniasis. J Liposome Res, 22:62–71.
- Rojo F, Albanell J, Rovira A, Corominas JM, Manzarbeitia F. 2008. Targeted therapies in breast cancer. Semin Diagn Pathol. 25: 245–261.
- Saul JM, Annapragada A, Natarajan JV, Bellamkonda RV. 2003. Controlled targeting of liposomal doxorubicin via the folate receptor in vitro. J Control Release. 92:49–67.
- Vyas SP, Katare YK, Mishra V, Sihorkar V. 2000. Ligand directed macrophage targeting of amphotericin B loaded liposomes. Int J Pharm. 210:1–14.
- Vyas SP, Quraishi S, Gupta S, Jaganathan KS. 2005. Aerosolized liposome-based delivery of amphotericin B to alveolar macrophages. Int J Pharm. 296:12–25.
- Vyas SP, Subhedar R, Jain S. 2006. Development and characterization of emulsomes for sustained and targeted delivery of an antiviral agent to liver. J Pharm Pharmacol. 58:321–326.
- Vyas SP, Sihorkar V, Jain S. 2007. Mannosylated liposomes for Bio-film targeting. Int J Pharm. 330:6–13.
- Xiang G, Wu J, Lu Y, Liu Z, Lee RJ. 2008. Synthesis and evaluation of a novel ligand for folate-mediated targeting liposomes. Int J Pharm. 356:29–36.
- Yuan Z, Chen D, Zhang S, Zheng Z. 2010. Preparation, characterization and evaluation of docetexal-loaded, folate conjugated PEG- liposomes. Yakugaku Zasshi. 130:1353–1359.