
Abstract
Objective: The present study traces the development and characterization of the gel formulation of valsartan-loaded ultradeformable vesicles for management of hypertension. Materials and methods: The prepared gel formulation of ultradeformable vesicles was evaluated for in vitro skin permeation, release kinetics, skin irritation, pharmacokinetics, and stability. Results and discussion: The in vitro skin permeation study showed that the gel formulation of ultradeformable vesicles presented a flux value of 368.74 μg/cm2/h, in comparison to that of the traditional liposomal gel formulation, with an enhancement ratio of 26.91, through rat skin. The data for release kinetics showed that the release profile followed zero-order kinetics, and that the drug release mechanism was non-Fickian. The results of the skin irritation study demonstrated that the prepared formulation was safe, less irritant, and well-tolerated for transdermal delivery. The results of the pharmacokinetic study demonstrated that the AUC value of valsartan after transdermal administration was apparently increased. The formulation stored under a refrigerated condition showed greater stability, and results were found to be within the specification under storage conditions. Conclusion: It is evident from this study that the gel formulation of ultradeformable vesicles of valsartan is a promising delivery system for lipophilic drugs, and has reasonably good stability characteristics.
Introduction
The major hurdle in transdermal delivery is the obstructive property of the stratum corneum, making this route of administration frequently unsuitable for medical use (CitationAhad et al. 2010, CitationAqil et al. 2007). Many approaches have been tried to overcome the skin's barrier property, and these include the use of chemical permeation enhancers, iontophoresis, microneedles, ultrasound, and vesicular systems such as liposomes, ethosomes, and ultradeformable vesicles (CitationAhad et al. 2009, CitationAhad et al. 2010, CitationAlexander et al. 2012, CitationElsabahy and Foldvari 2013). The conventionally used liposomes remain confined to the upper surface, with little penetration into the stratum corneum (CitationAhad et al. 2013, CitationAhad et al. 2014d). Hence, a new class of liposomes, also called elastic liposomes, transfersomes, or ultradeformable liposomes, have been developed. Ultradeformable vesicles are promising carriers for non-invasive transdermal delivery (CitationHiruta et al. 2006). Ultradeformable vesicles are prepared from phospholipids and edge activators. An edge activator is often a single-chain surfactant that destabilizes the lipid bilayers of the vesicles and increases the deformability of the bilayers. Sodium cholate, sodium deoxycholate, Span 60, Tween 20, and Tween 80 are some examples of edge activators which provide stress-dependent adaptability to these vesicular carriers so that they can easily squeeze between the pores of the stratum corneum. Compared with subcutaneous administration, the use of ultradeformable vesicles was found to improve in vitro skin permeation of various drugs, penetrate intact skin in vivo, and efficiently transfer therapeutic amounts of drugs (CitationCevc et al. 1998, CitationElsayed et al. 2006). The high deformability and hydration driving force of the ultradeformable vesicles both allowed and prompted the drug-loaded vesicles to move across the skin barrier (CitationCevc and Gebauer 2003).
In addition, ultradeformable vesicles are colloidal carriers which are easily accumulated in the leaky synovial tissue, which enables peripheral targeting. Ultradeformable vesicles also act as depots, enabling a controlled drug delivery system. Besides the advantages of their good affinity with skin, innocuity, and safety, the priorities associated with elastic liposomes were their high flexibility and high skin penetration (CitationEl Zaafarany et al. 2010).
Valsartan is used for the treatment of hypertension. It may be used alone or in combination with other antihypertensive agents. Valsartan is generally given for a longer duration of time. This requires a daily dosing schedule, which inconveniences the patient. The oral bioavailability of valsartan is approximately 25%. The low bioavailability is primarily due to incomplete absorption and partly due to pre-systemic metabolism. Peak plasma concentrations of valsartan occur 2 to 4 h after an oral dose (CitationAhad et al. 2014b, CitationAhad et al. 2013). The elimination half-life of valsartan is about 5 to 9 h. About 83% of valsartan is excreted in the feces and 13% in urine, following an oral dose (CitationAhad et al. 2014c, CitationAhad et al. 2011b). Due to the low bioavailability of valsartan after oral administration, and the inconveniences related to parenteral administration, the development of a transdermal system of valsartan administration is reasonably important (CitationAhad et al. 2014a).
From our laboratory, we have already reported the development, characterization, and in vitro permeation pattern of a transdermal system of administering valsartan using transfersomes (CitationAhad et al. 2012). The objective of the present investigation is to assess the release kinetics, stability studies, skin irritation, and in vivo pharmacokinetics of the gel system of valsartan-loaded ultradeformable vesicles in rats.
Materials and methods
Materials
Valsartan was received as a gratis sample from Ranbaxy Research Laboratories Ltd., Gurgaon, India. High Performance Liquid Chromatography (HPLC)-grade acetonitrile and methanol were purchased from Spectrochem Pvt. Ltd., India. Carbopol® 940, polyethylene glycol-400, orthophosphoric acid, and triethanolamine were purchased from S.D. Fine Chemicals, India. Potassium dihydrogen orthophosphate was purchased from Merck India Ltd., India. Water for HPLC was purchased from Thomas Baker (Chemicals) Ltd., Mumbai. Double-distilled water was used for all experiments.
Animals
Albino Wistar rats (200 g) were supplied by the Central Animal House of Jamia Hamdard University and housed under standard laboratory conditions in a 12 h light/dark cycle at 25 ± 2o C. The animals were nourished with a pellet diet (Lipton, India) and water ad libitum. The animals were received after the study was duly approved by the University Animal Ethics Committee, and by the CPSCEA (Committee for the Purpose of Control and Supervision of Experiments on Animals), Government of India.
Preparation of gel formulation of valsartan-loaded ultradeformable vesicles
Formulations of valsartan-loaded ultradeformable vesicles were previously prepared using the Box–Behnken design, by the conventional thin layer evaporation technique (CitationAhad et al. 2012). Briefly, Phospholipon® 90 G, surfactant, and the drug were taken in a clean, dry, round bottom flask, and the lipid mixture was dissolved in chloroform/methanol (2/1, v/v). The organic solvent was removed by rotary evaporation. The deposited lipid film was hydrated in a phosphate buffer solution by rotation at 60 rpm for 1 h at room temperature. To prepare smaller vesicles, large multilamellar vesicles were probe-sonicated, and these sonicated vesicles were then extruded through a sandwich of 200 and 450 nm polycarbonate membranes. Further, the valsartan-loaded ultradeformable vesicles were converted into a gel. Carbopol® 940 (1% w/w) was added into the water and kept overnight for complete humectation of the polymer chains. The formulation of valsartan ultradeformable vesicles was added to the hydrated Carbopol solution, with stirring. Other ingredients like polyethylene glycol-400 (15% w/v) and triethanolamine (0.5% w/v) were added to get a homogeneous dispersion of gel, and this gel formulation was used for the in vivo pharmacokinetic study.
Studies of in vitro skin permeation across rat skin
In vitro skin permeation studies of the valsartan-loaded ultradeformable vesicles gel system were carried out by studying permeation through rat skin, using the transdermal diffusion cell sampling system (SFDC 6, LOGAN Instruments, NJ, USA). The temperature of the receiver compartment was warmed with the built-in heater, thermostated at 37 ± 1°C throughout the experiments to provide an approximate skin surface temperature (i.e., 32 ± 1°C) (CitationAhad et al. 2011a, CitationMoghimi et al. 2009, CitationMura et al. 2013, CitationMura et al. 2009). A Teflon-coated mini magnetic bead was kept in the receiver compartment for agitating the contained vehicle at 600 rpm. The rat skin sample was mounted between the two diffusion cell compartments, with the stratum corneum facing toward the donor compartment (CitationJain et al. 2008, CitationRizwan et al. 2008). One gram of gel was placed in the donor compartment and covered with parafilm to avoid evaporation of water. The receptor aliquot samples (500 μL) were collected at predetermined time intervals and immediately replenished with the same amount of fresh vehicle. All the samples were analyzed by HPLC (CitationTatar and Saglik 2002).
Gel viscosity measurement
The viscosity measurements for the valsartan-loaded ultradeformable vesicles gel were determined at 25 ± 2°C, using a Brookfield R/S Plus cone and plate rheometer with spindle C50-1(Brookfield Engineering Laboratories Inc., Middleboro, MA, USA), at 50 rpm. All viscosity measurements were performed in triplicate (CitationAhad et al. 2014b, CitationChaudhary et al. 2013).
Drug content
For determination of drug content, about 1 g of the ultradeformable vesicles gel was weighed in a 100 ml volumetric flask and dissolved in methanol; it was diluted appropriately and analyzed by the HPLC method (CitationTatar and Saglik 2002). The content of valsartan in each sample was determined in terms of percentage of drug content.
pH evaluation
One hundred milligrams of the gel containing valsartan-loaded ultradeformable vesicles was weighed in a 50 mL volumetric flask, and then the volume was made up to 50 mL with double-distilled water (0.2% strength). The pH of the gel was recorded with a glass microelectrode (Mettler Instruments, Giessen, Germany) by bringing it in contact with the gel and allowing it to equilibrate for 1 min. Experiments were performed in triplicate, to check for the neutralization of gels of different batches (CitationChaudhary et al. 2011).
Extrudability, homogeneity, and spreadability
A simple method was adopted for the determination of extrudability in terms of grams required to extrude a 0.5 cm ribbon of gel in 10 s from a collapsible tube. The homogeneity of the gels was tested by visual inspection.
One of the criteria for a gel to meet the ideal qualities is that it should possess good spreadability. Spreadability is the term expressed to denote the extent of area to which the gel readily spreads on application to the skin or affected part. The therapeutic efficacy of a formulation also depends upon its spreadability value. The spreadability of the gel was determined using the following technique: 0.5 g of gel was placed within a circle of 2 cm diameter premarked on a glass plate, over which a second glass plate was placed. A weight of 500 g was allowed to rest on the upper glass plate for 5 min (CitationBachhav and Patravale 2009). The increase in the diameter due to spreading of the gel was noted. The percent spread by area was calculated using the following equation:
% Spread by area = (A2/A1)*100
where, A1 = 2 cm, and A2 = final area after spreading.
Release kinetics
To study the release kinetics, data obtained from in vitro permeation studies were fitted in various kinetic models (): zero order, as the cumulative percent of drug permeated vs time, first order, as the log cumulative percentage of drug remaining vs time, and Higuchi's model, as the cumulative percent drug permeated vs square root of time. To determine the mechanism of drug release, the data were fitted into the Korsmeyer–Peppas model as the log cumulative percentage of drug released vs log time, and the exponent n was calculated from the slope of the straight line. For the slab matrix, if the exponent is 0.5, then the diffusion mechanism is Fickian; if 0.5 < n < 1.0, the mechanism is non- Fickian, n = 1 indicates case II (relaxation) transport, and n > 1 indicates super case II transport (CitationGupta et al. 2012).
Table I. Mathematical models used to study the mechanism of release kinetics.
Skin irritation study
The skin irritation study was performed according to the method reported by Draize et al., using Wistar rats as the animal models (CitationDraize et al. 1944). The rats were divided into 2 groups. Group 1 received the gel formulation of the valsartan-loaded ultradeformable vesicles, while Group 2 received a 0.8% v/v aqueous solution of formalin as a standard irritant (positive control). Hair from the rats’ abdominal region was shaved using an electric clipper, approximately 24 h before the application of test formulation. Care was taken to avoid abrading the skin.
Fresh formulation and formalin solution (0.5 g) were applied on the hair-free abdominal side of the rats, for 7 days. After removal of the gel, the animals were examined for signs of erythema and edema, and the responses scored at days 3, 7, 24, and 48. An adjacent area of untreated skin of each animal was used as a control for the test. Dermal irritation was scored and recorded according to a visual scoring scale. The observations were given scores ranging from 0–4, depending on the degree of erythema and edema, as follows: no erythema 0, slight erythema (light pink) 1, moderate erythema (dark pink) 2, moderate to severe erythema (light red) 3, and severe erythema (extreme redness) 4 (CitationAqil et al. 2004, CitationDraize et al. 1944, CitationUbaidulla et al. 2007).
In vivo pharmacokinetic study
Wistar rats were used as the animal models for bioavailability studies. The animals were supplied by the Central Animal House facility of Jamia Hamdard, after approval was obtained from the Institutional Animal Ethics Committee, Jamia Hamdard, New Delhi (vide application form no. 532), and from the CPSEA. All the rats were kept under standard laboratory conditions in a 12-hour light/dark cycle at 25°C ± 2°C, and provided with a pellet diet (Lipton, Kolkata, India) and water ad libitum. The rodents’ skin was shaved on the abdominal side. Before application of the formulation, rats were kept under observation for 24 h, to detect any untoward effects of shaving; they were fasted over this period. The dose (3.6 mg) for the rats was calculated based on the weight of the rats according to the surface area ratio (CitationPaget and Barnes 1964). The valsartan ultradeformable vesicles gel was applied on each rat, and the blood samples were withdrawn at different time intervals (0, 2, 4, 6, 8, 12, 16, 20, 24, 36, and 48 h). The rats were anesthetized using diethyl ether, and blood samples (0.3 mL) were collected in Eppendorf tubes containing 8 mg of disodium EDTA as an anticoagulant. The blood samples collected were properly mixed with the anticoagulant, and plasma samples were separated by centrifugation (Remi Centrifuge, Mumbai, India) at 5000 rpm for 20 min and stored at –80°C until they were analyzed by HPLC (CitationPiao et al. 2008).
The pharmacokinetic parameters such as Cmax (maximum plasma concentration), time to reach maximum concentration (Tmax), and area under the curve from 0 to t (AUC0–48) and from 0 to ∞ (AUC0–∞) were calculated with the help of WinNonlin (version 5.2) software (Pharsight, Mountain View, CA, USA). The elimination rate constant (Kel) was calculated as the slope of the terminal portion of the log (plasma concentration) vs the time curve. The elimination half-life (t1/2) was calculated using the pharmacokinetic relationship
t1/2 = log 2/Kel.
Stability study of valsartan ultradeformable vesicles gel system
The gel system of the valsartan-loaded ultradeformable vesicles was subjected to stability studies to evaluate any physical or chemical changes on storage. For the stability study, the gel was kept at 4 ± 2°C for 6 months in a Borosil glass container (CitationGarg et al. 2010). The formulation samples were evaluated for clarity, pH, homogeneity, and drug content at 0, 1, 3 and 6 months after storage (CitationDave et al. 2010, CitationDuangjit et al. 2013).
Results and discussion
In vitro skin permeation studies of the gel across rat skin were performed. The in vitro permeation profile of the gel shows that the valsartan-loaded ultradeformable vesicles gel system presented the flux value of 368.74 ± 16.74 μg/cm2/h in comparison to the rigid liposomal gel formulation (13.70 ± 2.43 μg/cm2/h), with an enhancement ratio of 26.91 for permeation through rat skin. The reason for this better performance of the ultradeformable vesicles gel formulation is the flexibility and the ability of the vesicles to to retain their integrity, while the aggregates undergo a dramatic change in shape in comparison with those of conventional liposomes. All these characteristics allow the ultradeformable vesicles gel system to pass through the skin's pores, which are much smaller than their own diameter (CitationCevc and Blume 1992).
The prepared gel formulation was evaluated for cosmetic qualities such as color, scent, texture, and consistency. The valsartan ultradeformable vesicles gel prepared was white in color, with a pleasant, smooth, and homogeneous appearance and texture. The pH value of the prepared formulation was found to be 6.8, which is considered acceptable to avoid the risk of irritation upon application to the skin. It was found to be clear, free from the presence of particles, and showed good homogeneity, with an absence of lumps. The viscosity was found to be 34.96 ± 2.36 PaS. The spreadability of a gel plays a significant role in patient compliance and helps in uniform application of gel to the skin. A good gel takes less time to spread and will have high spreadability. The developed gel of valsartan ultradeformable vesicles presented a spreadability of 336.52%, with excellent extrudability, that is, the gel was easily spreadable. The drug content of the gel formulation was found to be 99.98%, showing content uniformity. In order to propose the possible pattern of drug release from the valsartan ultradeformable vesicles gel, the release was evaluated to check the goodness of fit for zero-order release kinetics, first-order release kinetics, Higuchi’s square root of time equation (CitationHiguchi 1963), and Korsmeyer–Peppas` power law equation (CitationKorsmeyer et al. 1983, CitationPeppas 1985). The goodness of fit was evaluated by R2 (correlation coefficient) values. The model showing the highest value of R2 was considered as the best model for release kinetics. The drug release from the gel system through rat skin was fitted into different mathematical models, and the correlation coefficient was found. The R2 values for different models are presented in , for gel permeation across rat skin. The plot of cumulative drug release across rat skin fitted to different models of kinetics is presented in . The correlation coefficients obtained after fitting the in vitro permeation data to the respective model equations indicate that the best fit is obtained with zero order for the valsartan-loaded ultradeformable vesicles gel. The correlation coefficient obtained for the valsartan-loaded ultradeformable vesicles gel system was 0.9937 (zero order) for data on permeation across rat skin.
Figure 1. The plots of the kinetics of drug release from the valsartan ultradeformable vesicles gel through rat skin, fitted to (A) zero order, (B) first order, (C) Higuchi, and (D) Peppas’ mathematical kinetics model.
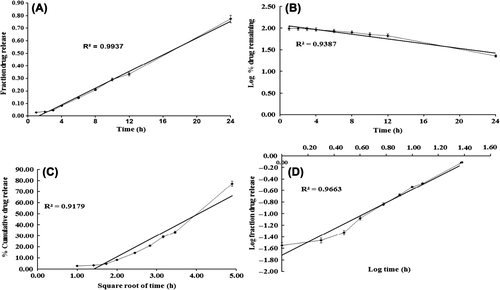
Table II. Correlation coefficients found by fitting data to different mathematical models for permeation of the valsartan ultradeformable vesicles gel across rat skin.
The zero-order release model ideally describes the release of the modified-release dosage form, including transdermal delivery systems. In the present study, the ultradeformable vesicles gel system showed zero-order release kinetics. The same release kinetics, for permeation of transfersomal and ethosomal gels across the skin membrane, was described by previous researchers (CitationBendas and Tadros 2007, CitationGarg et al. 2010, CitationGlavas-dodov et al. 2003). The pharmaceutical dosage forms following this profile release the same amount of drug by unit of time, and it is the ideal method of drug release to achieve a prolonged pharmacological action (CitationCosta and Sousa Lobo 2001).
Ritger and Peppas introduced the power law equation, mt/m∞ = ktn, to characterize the controlled-release behavior of a drug from polymer matrices (CitationRitger and Peppas 1987). The value of slope (n) of log mt/m∞ vs log t, is the release exponent, which indicates the operating release mechanism. Peppas used this n value in order to characterize different release mechanisms, concluding for the values for a slab, of n ≤ 0.5 for Fickian diffusion, and higher values of n, between 0.5 and 1.0, or n = 1.0, for mass transfer, following the non-Fickian model (CitationPeppas 1985, CitationRitger and Peppas 1987).
Based on the high R2 value, it is clear that the release of valsartan from the ultradeformable vesicles gel system follows zero-order kinetics. Since the value of the release exponent for the proposed model was greater than 1 (n > 1) in the case of the ultradeformable vesicles gel system developed, it was found that the release mechanism predominating the permeation of the gel system across rat skin was a non-Fickian super case II release mechanism.
The test formulation (valsartan ultradeformable vesicles gel system) was evaluated for primary skin irritation, in accordance with the method reported by Draize and coworkers (CitationDraize et al. 1944). The purpose of this test was to ascertain the dermal irritation potential of the test formulation on the intact rat skin. After applications of test formulation and standard irritant, the application sites were examined for dermal reactions, in accordance with Draize scoring criteria. The primary irritation score was calculated following the completion of the study. The result of the skin irritation test for the gel system of valsartan-loaded ultradeformable vesicles after application is reported in . According to Draize et al., compounds producing scores of 2 or less are considered negative, that is, they cause no skin irritation (CitationDraize et al. 1944).
Table III. Average response of skin irritation scores following multiple applications of the valsartan ultradeformable vesicles gel system and formalin on Wistar rat skin for a period of 7 days.
The mean irritation scores of erythema and edema on application of the ultradeformable vesicles gel were found to be 1.0 and 0.6 respectively. On the other hand, application of the standard irritant, formalin, produced irritation scores of 2.4 and 2.2 for erythema and edema respectively. In light of the above observations, it was concluded that the ultradeformable vesicles gel system developed was a safe, less irritant, and well-tolerated formulation for transdermal delivery.
The plasma concentration profile of valsartan following the application of the ultradeformable vesicles gel system in rats is shown in .
Figure 2. Plasma concentration profile of valsartan after transdermal application of the valsartan ultradeformable vesicles gel system in rats.
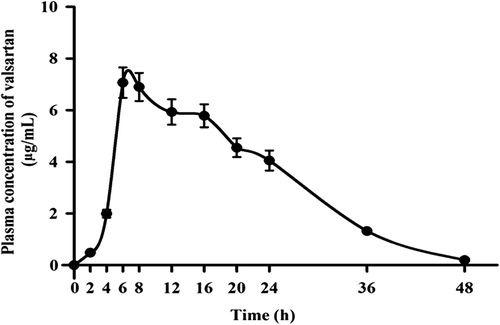
The Cmax, after application of the ultradeformable vesicles gel, was found to be 7.07 ± 0.06 μg/ml, and Tmax was 5.5 ± 0.67 h.
CitationKim and Baek (2014) reported a Tmax value of around 1.9 h after oral administration of valsartan (CitationKim and Baek 2014). This higher value of Tmax in our study, about 5.5 h after transdermal application, was because of the barrier property of the stratum corneum (CitationRizwan et al. 2008). A Cmax of 7280 ± 979 ng/mL on transdermal administration of valsartan in hairless rats has been reported (CitationNishida et al. 2010). It was also observed that the values for AUC0–48, and AUC0-∞ of the gel formulation were 154.06 ± 7.56 μg/ml/h and 165.60 ± 10.93 μg/ml/h2 respectively. The AUC value obtained after the application of the valsartan ultradeformable vesicles gel system could be due to the avoidance of hepatic metabolism by transdermal administration, and indicated high systemic drug availability. The t1/2 value of 40.40 ± 2.01 h of valsartan in plasma was noticed after the application of the ultradeformable vesicles gel system. This may be caused by the longer duration of absorption via the transdermal system. In summary, the ultradeformable vesicles gel system developed has provided better bioavailability of drug, and could provide a useful strategy for better management of hypertension.
To assess the influence of storage condition on the physical stability of the ultradeformable vesicles gel system, stability study was performed in adherence to the ICH guidelines. The vesicles gel system was stored at 4 ± 2°C for stability, for a period of 6 months. The samples were periodically withdrawn at predetermined time intervals, during the initial phase, 1, 3, and 6 months, and evaluated for various parameters. The results of the parameter study are presented in .
Table IV. The assessment of stability of the ultradeformable vesicles gel system, according to the ICH guidelines, under conditions of 4 ± 2°C for six months.
The vesicles gel system remained clear and homogenous; no phase separation was observed on storage for 6 months. It is suggested that the vesicles gel system should be maintained under refrigerated conditions, to minimize the drug leakage (CitationAgarwal et al. 2001). The drug leakage of less than 5% of the initial load under conditions of elevated temperature and refrigeration is well within the acceptable limits, when vesicles are to be advocated for topical applications (CitationPatel et al. 2010).
Conclusion
These findings indicate that the transdermal gel containing ultradeformable vesicles may offer promise as an effective dosage form for antihypertensive drugs, ensuring more effective therapy. Further, incorporating the vesicles formulation into the Carbopol-loaded transdermal gel formulation increases the stability of the liquid formulation, which is unaffected by aging and does not support bacterial or fungal growth. The resulting formulation has a pleasant texture and is nonirritating.
Conflict of interest
All authors have approved the final manuscript, and the authors declare that they have no conflicts of interest to disclose.
References
- Agarwal R, Katare OP, Vyas SP. 2001. Preparation and in vitro evaluation of liposomal/niosomal delivery systems for antipsoriatic drug dithranol. Int J Pharm. 228:43–52.
- Ahad A, Al-Mohizea AM, Al-Jenoobi FI, Aqil M. 2014a. Transdermal delivery of angiotensin II receptor blockers (ARBs), angiotensin-converting enzyme inhibitors (ACEIs) and others for management of hypertension. Drug Deliv, Posted online on July. 28:1–12.
- Ahad A, Aqil M, Ali A. 2014b. Investigation of antihypertensive activity of carbopol valsartan transdermal gel containing 1,8-cineole. Int J Biol Macromol. 64:144–149.
- Ahad A, Aqil M, Kohli K, Chaudhary H, Sultana Y, Mujeeb M, Talegaonkar S. 2009. Chemical penetration enhancers: a patent review. Expert Opin Ther Pat. 19:969–988.
- Ahad A, Aqil M, Kohli K, Sultana Y, Mujeeb M. 2013. Enhanced transdermal delivery of an anti-hypertensive agent via nanoethosomes: statistical optimization, characterization and pharmacokinetic assessment. Int J Pharm. 443:26–38.
- Ahad A, Aqil M, Kohli K, Sultana Y, Mujeeb M. 2014c. Design, formulation and optimization of valsartan transdermal gel containing iso-eucalyptol as novel permeation enhancer: Preclinical assessment of pharmacokinetic in wistar albino rats. Expert Opin Drug Deliv. 11:1149–1162.
- Ahad A, Aqil M, Kohli K, Sultana Y, Mujeeb M, Ali A. 2010. Transdermal drug delivery: the inherent challenges and technological advancements. Asian J Pharm Sci. 5:276–288.
- Ahad A, Aqil M, Kohli K, Sultana Y, Mujeeb M, Ali A. 2011a. Interactions between novel terpenes and main components of rat and human skin: mechanistic view for transdermal delivery of propranolol hydrochloride. Curr Drug Deliv. 8:213–224.
- Ahad A, Aqil M, Kohli K, Sultana Y, Mujeeb M, Ali A. 2011b. Role of novel terpenes in transcutaneous permeation of valsartan: effectiveness and mechanism of action. Drug Dev Ind Pharm. 37:583–596.
- Ahad A, Aqil M, Kohli K, Sultana Y, Mujeeb M, Ali A. 2012. Formulation and optimization of nanotransfersomes using experimental design technique for accentuated transdermal delivery of valsartan. Nanomedicine. 8:237–249.
- Ahad A, Raish M, Al-Mohizea AM, Al-Jenoobi FI, Alam MA. 2014d. Enhanced anti-inflammatory activity of carbopol loaded meloxicam nanoethosomes gel. Int J Biol Macromol. 67:99–104.
- Alexander A, Dwivedi S, Ajazuddin, Giri TK, Saraf S, Tripathi DK. 2012. Approaches for breaking the barriers of drug permeation through transdermal drug delivery. J Control Release. 164:26–40.
- Aqil M, Ahad A, Sultana Y, Ali A. 2007. Status of terpenes as skin penetration enhancers. Drug Discov Today. 12:1061–1067.
- Aqil M, Ali A, Sultana Y, Parvez N. 2004. Communication: Matrix type transdermal drug delivery systems of metoprolol tartrate: skin toxicity and in vivo characterization. Ethiop Pharm J. 22:53–60.
- Bachhav YG, Patravale VB. 2009. Microemulsion based vaginal gel of fluconazole: formulation, in vitro and in vivo evaluation. Int J Pharm. 365:175–179.
- Bendas ER, Tadros MI. 2007. Enhanced transdermal delivery of salbutamol sulfate via ethosomes. AAPS PharmSciTech. 8:E107.
- Cevc G, Blume G. 1992. Lipid vesicles penetrate into intact skin owing to the transdermal osmotic gradients and hydration force. Biochim Biophys Acta. 1104:226–232.
- Cevc G, Gebauer D. 2003. Hydration-driven transport of deformable lipid vesicles through fine pores and the skin barrier. Biophys J. 84:1010–1024.
- Cevc G, Gebauer D, Stieber J, Schatzlein A, Blume G. 1998. Ultraflexible vesicles, Transfersomes, have an extremely low pore penetration resistance and transport therapeutic amounts of insulin across the intact mammalian skin. Biochim Biophys Acta. 1368: 201–215.
- Chaudhary H, Kohli K, Amin S, Rathee P, Kumar V. 2011. Optimization and formulation design of gels of Diclofenac and Curcumin for transdermal drug delivery by Box-Behnken statistical design. J Pharm Sci. 100:580–593.
- Chaudhary H, Rohilla A, Rathee P, Kumar V. 2013. Optimization and formulation design of carbopol loaded Piroxicam gel using novel penetration enhancers. Int J Biol Macromol. 55:246–253.
- Costa P, Sousa Lobo JM. 2001. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 13:123–133.
- Dave V, Kumar D, Lewis S, Paliwal S. 2010. Ethosome for Enhanced Transdermal Drug Delivery of Aceclofenac. Int J Drug Deliv. 2: 81–92.
- Draize JH, Woodard G, Calvery HO. 1944. Methods for the study of irritation and toxicity of substances applied topically to the skin and mucous membranes. J Pharmacol Exp Ther. 82:377–390.
- Duangjit S, Opanasopit P, Rojanarata T, Ngawhirunpat T. 2013. Evaluation of meloxicam-loaded cationic transfersomes as transdermal drug delivery carriers. AAPS PharmSciTech. 14:133–140.
- El Zaafarany GM, Awad GA, Holayel SM, Mortada ND. 2010. Role of edge activators and surface charge in developing ultradeformable vesicles with enhanced skin delivery. Int J Pharm. 397: 164–172.
- Elsabahy M, Foldvari M. 2013. Needle-free gene delivery through the skin: an overview of recent strategies. Curr Pharm Des. 19:7301–7315.
- Elsayed MM, Abdallah OY, Naggar VF, Khalafallah NM. 2006. Deformable liposomes and ethosomes: mechanism of enhanced skin delivery. Int J Pharm. 322:60–66.
- Garg AK, Mohan NL, Meenakshi C. 2010. Gel containing ethosomal vesicles for transdermal delivery of aceclofenac. Int J Pharmacy Pharm Sci. 2:102–108.
- Glavas-dodov M, Fredro-kumbaradzi E, Goracinova K, Calis S, Simonoska M, Hincal AA. 2003. 5-Fluorouracil in topical liposome gels for anticancer treatment -Formulation and evaluation. Acta Pharmacol Sin. 53:241–245.
- Gupta A, Aggarwal G, Singla S, Arora R. 2012. Transfersomes: a novel vesicular carrier for enhanced transdermal delivery of sertraline: development, characterization, and performance evaluation. Sci Pharm. 80:1061–1080.
- Higuchi T. 1963. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 52:1145–1149.
- Hiruta Y, Hattori Y, Kawano K, Obata Y, Maitani Y. 2006. Novel ultra-deformable vesicles entrapped with bleomycin and enhanced to penetrate rat skin. J Control Release. 113:146–154.
- Jain R, Aqil M, Ahad A, Ali A, Khar RK. 2008. Basil oil is a promising skin penetration enhancer for transdermal delivery of labetolol hydrochloride. Drug Dev Ind Pharm. 34:384–389.
- Kim MS, Baek IH. 2014. Fabrication and evaluation of valsartan-polymer- surfactant composite nanoparticles by using the supercritical antisolvent process. Int J Nanomedicine. 9:5167–5176.
- Korsmeyer RW, Gurny R, Doelker EM, Buri P, Peppas NA. 1983. Mechanism of solute release from porous hydrophilic polymers. Int J Pharm. 15:25–35.
- Moghimi HR, Makhmalzadeh BS, Manafi A. 2009. Enhancement effect of terpenes on silver sulphadiazine permeation through third-degree burn eschar. Burns. 35:1165–1170.
- Mura S, Manconi M, Fadda AM, Sala MC, Perricci J, Pini E, Sinico C. 2013. Penetration enhancer-containing vesicles (PEVs) as carriers for cutaneous delivery of minoxidil: in vitro evaluation of drug permeation by infrared spectroscopy. Pharm Dev Technol. 18:1339–1345.
- Mura S, Manconi M, Sinico C, Valenti D, Fadda AM. 2009. Penetration enhancer-containing vesicles (PEVs) as carriers for cutaneous delivery of minoxidil. Int J Pharm. 380:72–79.
- Nishida N, Taniyama K, Sawabe T, Manome Y. 2010. Development and evaluation of a monolithic drug-in-adhesive patch for valsartan. Int J Pharm. 402:103–109.
- Paget GM, Barnes JM. 1964. Interspecies dosage conversion scheme in evaluation of results and quantitative application in different species. In: Laurence DR, Bacharach AL, Eds. Evaluation of Drug Activities: Pharmacometrics. London and New York: Academic press. pp. 160–162.
- Patel SS, Patel MS, Salampure S, Vishwanath B, Patel NM. 2010. Development and evaluation of liposomes for topical delivery of tacrolimus (Fk-506). J Sci Res. 2:585–596.
- Peppas NA. 1985. Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv. 60:110–111.
- Piao ZZ, Lee ES, Tran HT, Lee BJ. 2008. Improved analytical validation and pharmacokinetics of valsartan using HPLC with UV detection. Arch Pharm Res. 31:1055–1059.
- Ritger PL, Peppas NA. 1987. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Control Release. 5:23–36.
- Rizwan M, Aqil M, Ahad A, Sultana Y, Ali MM. 2008. Transdermal delivery of valsartan: I. Effect of various terpenes. Drug Dev Ind Pharm. 34:618–626.
- Tatar S, Saglik S. 2002. Comparison of UV- and second derivative-spectrophotometric and LC methods for the determination of valsartan in pharmaceutical formulation. J Pharm Biomed Anal. 30:371–375.
- Ubaidulla U, Reddy MV, Ruckmani K, Ahmad FJ, Khar RK. 2007. Transdermal therapeutic system of carvedilol: effect of hydrophilic and hydrophobic matrix on in vitro and in vivo characteristics. AAPS PharmSciTech. 8:2.