Abstract
Alterations in gene expression are implicated in the pathogenesis of several neuropsychiatrie disorders, including drug addiction and depression, increasing evidence indicates that changes in gene expression in neurons, in the context of animal models of addiction and depression, are mediated in part by epigenetic mechanisms that alter chromatin structure on specific gene promoters. This review discusses recent findings from behavioral, molecular, and bioinformatic approaches that are being used to understand the complex epigenetic regulation of gene expression in brain by drugs of abuse and by stress. These advances promise to open up new avenues for improved treatments of these disorders.
Las alteraciones en la expresión génica están implicadas en la patogénesis de varios trastornos neuropsiquiátricos, incluyendo la adicción a drogas y la depresión. Existe una evidencia creciente que indica que los cambios en la expresión génica en las neuronas, en el contexto de modelos animales de adicción y depresión, están mediados en parte por mecanismos epigenéticos que alteran la estructura de la cromatina de genes promotores especificos. Esta revisión discute hallazgos recientes que provienen de aproximaciones conductuales, moleculares y bioinformáticas los cuales están siendo utilizados para comprender la compleja regulación epigenética de la expresión génica en el cerebro por las drogas de abuso y por el estrés. Estos avances prometen establecer nuevos caminos para mejores tratamientos de estos trastornos.
Des modifications de l'expression génique sont impliquées dans la pathogenèse de plusieurs maladies neuropsychiatriques, y compris la toxicomanie et la dépression. Des modèles animaux de ces pathologies montrent de plus en plus que des variations de l'expression des gènes dans les neurones sont transmises en partie par des mécanismes épigénétiques qui changent la structure de la chromatine sur les promoteurs spécifiques des gènes. Cet article analyse les résultats récents des approches comportementales, moléculaires et bioinformatiques utilisées pour comprendre la régulation épigénétique complexe de l'expression génique dans le cerveau par l'abus de substances et par le stress. Ces avancées ouvrent de nouvelles voies pour mieux traiter ces maladies.
Family history is one of the greatest risk factors for psychiatric disorders, yet their genetic basis remains poorly understood despite substantial advances in whole genome sequencing techniques. While the search for genetic mutations continues at a rapid pace, the field is also investigating the environmental component of family history, which has remained more difficult to explain mechanistically. One hypothesis is that environmental stimuli alter gene expression patterns in certain brain regions that ultimately change neural function and behavior. Support for this hypothesis has been observed in animal models of psychiatric illness, as well as in human patients.
The interactions between the environment and the genes that give rise to specific phenotypes are termed “epigenetic.”Citation1 An example of this process is observed in cellular differentiation, where unique chemical signals induce totipotent stem cells to differentiate into genetically identical cell types with vastly different functions. This is due in part to the vastly different sets of genes expressed between distinct cell types (eg, neurons vs hepatocytes), despite their identical DNA templates. Mechanistic insight into this process has recently been uncovered, and involves the transduction of unique environmental signals into precise and highly stable alterations in chromatin structure that ultimately gate access of transcriptional machinery to specific gene programs, thereby providing unique gene expression profiles in response to specific environmental cues.Citation2 Importantly, many of these chromatin remodeling mechanisms are highly stable, contributing to the maintenance of specific gene expression programs in the correct tissues throughout the life of an individual.
The strong control exerted by chromatin remodeling on gene expression, and the potential stability of chromatin mechanisms, make chromatin regulation a prime candidate for mediating aspects of the long-lasting neural plasticity that ultimately results in psychiatric syndromes. It is also interesting to note that certain neurological and psychiatric diseases are caused by rare genetic mutations in chromatin remodeling enzymes (Table I). While these mutations are rare, they directly illustrate how disruption of chromatin regulation can profoundly affect neural function and lead to complex behavioral abnormalities. Thus, epigenetic research in psychiatry is aimed at identifying whether environmental stimuli induce changes in chromatin structure which ultimately contribute to transcriptional programs in neurons that cause psychiatric illness, much in the same way as environmental cues differentiate a stem cell into specific lineages. While this field is still in its infancy, great progress is being made in identifying epigenetic alterations in many neuropsychiatric syndromes, including drug addiction, depression, schizophrenia, Alzheimer's disease, and Rett syndrome, among others. Focusing on drug addiction and depression, this review briefly discusses the molecular machinery underlying epigenetic mechanisms in brain, and how their dysregulation may contribute to these chronic psychiatric illnesses.
Table I. Examples of diseases of chromatin remodeling. CBP, CREB binding protein; CREB, cyclic AMP-response element binding protein; DMPK, DM1 protein kinase; Dnmt3B, DNA methyltransferase 3B; FMR1, fragile X mental retardation protein 1 ; MeCP2, methyl-CpG-binding protein 2; RSK2, ribosomal S6 kinase 2; SWI/SNF, mating switching and sucrose non-fermenting complex; UTR, untranslated region; XH2, X-linked helicase 2
Epigenetic mechanisms
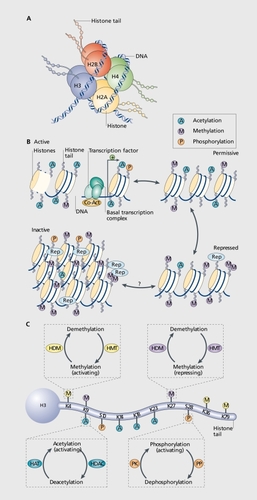
Chromatin is the complex of DNA, histories, and associated nonhistone proteins in the cell nucleus. DNA wraps around histone octamers made up of two copies of histone H2A, H2B, H3, and H4,Citation3 which then supercoil to form a highly condensed structure (). Initially, it was thought that this elaborate chromatin structure only functioned to condense meters of DNA into the microscopic cell nucleus, but it is now known to participate directly in gene regulation. Because DNA is tightly associated with histones and often embedded deep within chromatin supercoils,Citation4,Citation5 cellular mechanisms exist to modify and remodel chromatin structure to allow for the coordinated expression of specific transcriptional programs and the silencing of others. Citation6 Such modifications typically occur on N-terminal histone tails and include acetylation, phosphorylation, methylation, or several other covalent modifications of histones, methylation of DNA, and many others, with each modification either directly altering histone-DNA interactions or serving as a mark that recruits specific proteins to positively or negatively regulate the underlying gene's activity. Ultimately, dozens of potential modifications that occur at many distinct histone residues summate to determine the final transcriptional output of a given gene.Citation7 As mentioned earlier, genetic mutations in many of these chromatin remodeling enzymes are associated with severe neurological and psychiatric disorders (see Table I). An especially important aspect of certain chromatin modifications is their apparent stability, as is seen with genetic imprinting or X-inactivation, where DNA methylation contributes to lifelong gene silencing.Citation8 However, despite the apparent stability of some epigenetic mechanisms in vivo, all types of chromatin modifications identified to date are potentially reversible and have specific enzymes or processes which mediate the addition or removal of each mark.Citation6 The in vivo mechanisms which maintain easily reversible histone modifications (eg, acetylation) on some genes or delete highly stable marks (eg, DNA methylation) on other genes are currently not clear.
Histone acetylation
Acetylation of histone lysine residues reduces the electrostatic interaction between histone proteins and DNA, which relaxes chromatin structure and improves access of transcriptional regulators to DNA ().Citation6 Genome-wide studies indicate that high levels of histone acetylation in gene promoter regions are generally associated with higher gene activity, while low levels of acetylation correlating with reduced gene activity.Citation9 Most genome-wide studies of histone acetylation have focused on acetylation of the N-terminal lysine residues in histones H3 and H4, but histone acetylation can occur on other histone proteins as well as in their globular domains.
Histone acetylation is a dynamic process, controlled by specific enzymes which either add or remove the acetyl mark. There are over a dozen known histone acetyltransferases (HATs) which catalyze the addition of acetyl groups onto lysine residues of histones with varying degrees of specificity. Many HATs can also acetylate nonhistone proteins such as transcription factors (eg, p53), and some transcription factors (eg, ATF2 [activating transcription factor 2], CLOCK) even possess intrinsic HAT activity that contributes to gene activation.Citation10,Citation11
Histone deacetylases (HDACs), which remove acetyl groups from histones, are divided into four classes. Class I HDACs (eg, HDAC1, 2, 3, and 8) are ubiquitously expressed and likely mediate the majority of deacetylase activity within cells. Class II HDACs (eg, HDAC4, 5, 7, 9, 10) are only expressed in specific tissues such as heart and brain and are much larger enzymes that also contain an N-terminal regulatory domain that enables them to be shuttled in and out of the nucleus in a neural activitydependent manner.Citation12 While Class II HDACs can deacetylate histones, they are much less efficient enzymes than Class I HDACs, and may also deacetylate other cellular substrates.Citation13,Citation14 There is currently one Class IV HDAC, HDAC11, and it has characteristics of both Class I and Class II enzymes.Citation15 Class III IIDACs (also referred to as sirtuins) are mechanistically distinct from the other HDACs, and have been implicated in the regulation of lifespan and metabolism.Citation16 The individual functions of each IIDAC remain an active topic of investigation.
Histone phosphorylation
Histone phosphorylation is generally associated with transcriptional activation; it can be observed on the promoters of immediate early genes such as c-fos when they are induced after cyclic adenosine monophosphate (cAMP) stimulation or glutamate treatment in cultured striatal neurons.Citation17,Citation18 One of the best-characterized histone phosphorylation sites is serine 10 on histone H3 (H3S10).This modification stabilizes the HAT, GCN5, on associated gene promoters while antagonizing the repressive modification - methylation of lysine 9 on histone H3 (H3K9) and its subsequent recruitment of HP1 (heterochromatin protein 1, see below).Citation6 Since phosphorylation at H3S10 recruits a HAT, the neighboring lysine residue at H3K9 is often acetylated in concert with phosphorylation, a process called phosphoacetylation that further potentiates gene activation.
There are several nuclear protein kinases and protein phosphatases known to regulate histone phosphorylation.Citation6 The mitogen-activated protein kinase, MSK1, and the dopamine and cyclic-AMP regulated protein phosphatase inhibitor, DARRP-32, are elegant examples shown to regulate H3S10 phosphorylation in the adult brain in response to cocaine exposure.Citation19,Citation20 Furthermore, genetic disruption of the histone-modifying ability of MSK1 or DARRP-32 in vivo has dramatic effects on behavioral responses to cocaine. Thus, histone phosphorylation likely plays an important role in the regulation of brain function.
Histone methylation
Histone methylation generates unique docking sites that recruit transcriptional regulators to specific gene loci. Histone methylation occurs on lysine residues in mono-, di-, or trimethylated states, enabling each state to recruit unique coregulators and exert distinct effects on transcriptional activity.Citation6 Additionally, methylation of different histone lysine residues can exert opposite effects on transcription. In gene promoter regions for example, trimethylation of IT3K4 is highly associated with gene activation, whereas trimethylation of H3K9 or H3K27 is repressive.Citation5 The repression caused by trimethylation of II3K9 is mediated in part via the recruitment of corepressors, such as HP1, as stated earlier. However, even this is an oversimplification, as methylated H3K9 is often found in the coding region downstream of a gene promoter and may be involved in transcriptional elongation.Citation6,Citation21 Thus, histone methyiation provides each cell with exquisite control over an individual gene's activity through numerous combinatorial possibilities.
Histone methyltransferases (HMTs) add methyl groups to specific lysine residues of histones, and histone demelhylases (HDMs) remove them (). Like HATs and HDACs, HMTs and HDMs also have activity towards nonhistone proteins.Citation6 HMTs and HDMs not only discriminate between various histone lysine residues, but each enzyme is also unique in its ability to catalyze mono-, di-, or trimethylation or demethylation at that site.Citation6 For example, the HMT, KMT1C (G9a), is specific for histone H3K9 but only adds 1 or 2 methyl groups, with the distinct HMT, KMT1A (SUV39H1), catalyzing trimethylation of this site. Similarly, the HDM, KDM3A (JHDM2a), can demethylate 1 or 2 methyl groups on H3K9, requiring a distinct demethylase, eg, KDM4D (JMJD2D) to fully demethylate the trimethylated state. Thus, large complexes of enzymes are required to move between the unmethylated and fully trimethylated states. Proper balance of histone methylation has already been strongly implicated in normal brain function, as the HDM, KMT5C (SMCX), controls dendritic spine density and is mutated in patients with mental retardation.Citation22,Citation23
DNA methylation
DNA methylation refers to the enzymatic methylation of cytosine bases, a fundamental cellular process required for development, tissue-specific gene expression, X-inactivation, and genetic imprinting, to name a few examples.Citation24 DNA methylation is thought to repress gene expression by interfering with the binding of transcription factors to their target sequences or by initiating the recruitment of corepressors. For example, the cAMP-response element (CRE) contains a cytosine-guanine dinucleotide in the middle of its consensus sequence, which, when methylated, prevents the transcription factor CRE-binding protein (CREB) from binding.Citation25 Thus, for genes at which CREB is necessary to initiate transcription, methylation at this site is repressive. Methylated DNA can also recruit methyl-binding domain-containing proteins, such as MeCP2, which can then recruit and stabilize transcriptional corepressors such as HDACs on specific gene promoters. Mutations in MeCP2 cause the autistic spectrum disorder, Rett syndrome, illustrating the importance of DNA methylation in normal brain development.Citation26 While there is a strong correlation between methylated DNA and repressed gene activity, recent studies of MeCP2 indicate it may also serve to activate gene activity under some circumstances,Citation27 suggesting that the context in which DNA methylation occurs is an important factor in its ultimate effect on transcription.
There are three known enzymes which catalyze DNA cytosine methylation: DNMT1, DNMT3a, and DNMT3b. DMNT2 was recently shown to methylate RNA rather than DNA.Citation28 Together, these enzymes establish and maintain the unique methylation patterns that exist within each cell type. While the regulation of these enzymes in brain remains unclear, pharmacological inhibition of DNA methylation in the brain in vivo results in rapid demethylation of specific gene targets and severe deficits in learning and memory.Citation29 The mechanism by which this occurs, however, remains unclear because, unlike other chromatin modifications, the existence of DNA demethylases remains controversial.Citation30 Nevertheless, regulation of DNA methylation by environmental stimuli remains an attractive mediator of long-lasting changes in transcription in adult neurons.
Epigenetic mechanisms in drug addiction
Drug addiction is a chronic relapsing disorder where motivation to seek and take drugs of abuse becomes compulsive and pathological.Citation31 The process by which repeated drug experimentation transitions into a chronically addicted state is the focus of intense research, as clues into these mechanisms may help better manage or perhaps fully treat addicted patients. Another avenue of intense research focuses on the mechanisms driving drug relapse, which occurs even after long periods of drug abstinence and is a major clinical challenge for successful treatment. The exciting new possibility that druginduced alterations in chromatin structure may contribute to long-lasting behavioral changes provides a new avenue for novel therapeutics that improve drug rehabilitation.
The first studies to implicate changes in chromatin structure in responses to drugs of abuse found that acute administration of cocaine rapidly increased histone H4 acetylation on the immediate early genes c-fos and fosB in striatum,Citation32 two genes known to play a critical role in cocaine-related behaviors.Citation33 Hie histone acetyltransferase CBP appears to be required for the drug-induced acetylation of the fosB promoter, and probably many other, yet to be identified genes as well.Citation34 Interestingly, despite several control gene promoters where acute cocaine does not affect histone acetylation, an acute cocaine dose increases total levels of histone H4 acetylation, and histone H3 phosphoacetylation in striatum, as measured by Western blotting.Citation19,Citation32 These global increases in histone acetylation, which are also observed in response to environmental enrichment and tests of learning and memory,Citation35, Citation36 may be accounted for by high levels of acetylation on specific subsets of genes. This is likely, as global increases in histone K9 methylation, a repressive histone modification, are also observed after cocaine exposureCitation37 and appear to occur on unique subsets of genes.Citation38
The promoters of certain genes induced by chronic cocaine exposure are hyperacetylated for days to weeks after the last drug exposure (). For example, the expression of cdk5 (cyclin-dependent kinase 5), bdnf (brain derived neurotrophic factor),Citation37 npy (neuropeptide Y),Citation39 and sirt1 and sirt2 (two subtypes of sirtuins), among many other genes,Citation38 were found to be upregulated after chronic cocaine administration and their gene promoters hyperacetylated, while egr-1 (early growth response 1) was found to be downregulated and hypoacetylated after cocaine withdrawal.Citation39 Moreover, altered expression of each of these genes has been shown to contribute to the addiction behavioral phenotype. These findings suggest a role of histone acetylation in the maintenance of gene expression involved in drug addiction, including drug withdrawal and relapse.
![Figure 2. Regulation of chromatin structure by drugs of abuse. Drug-induced signaling events are depicted for psychostimulants such as cocaine and amphetamine. These drugs increase cAMP levels in striatum, which activates protein kinase A (PKA) and leads to phosphorylation of its targets. This includes the cAMP response element binding protein (CREB), the phosphorylation of which induces its association with the histone acetyltransferase, CREB binding protein (CBP) to acetylate histones and facilitate gene activation. This is known to occur on many genes including fosB and c-fos in response to psychostimulant exposure. AFosB is also upregulated by chronic psychostimulant treatments, and is known to activate certain genes (eg, cdk5) and repress others (eg, c-fos) where it recruits HDAC1 as a corepressor. This repression of c-fos also involves increased repressive histone methylation, which is thought to occur via the induction of specific histone methyltransferases (HMTs). In addition, cocaine regulates the HMT, KMT1 C/G9a, which alters histone H3 methylation on K9. It is not yet known how cocaine regulates histone demethylases (HDM) or DNA methyltransferases (DNMTs). Cocaine also activates the mitogen activated protein kinase (MAPK) cascade, which through MSK1 can phosphorylate CREB and histone H3 at serine 10. Cocaine promotes H3 phosphorylation via a distinct pathway, whereby PKA activates protein phosphatase 2A, leading to the dephosphorylation of serine 97 of DARPP32. This causes DARPP32 to accumulate in the nucleus and inhibit protein phosphatase-1 (PP1) which normally dephosphorylates H3. Chronic exposure to psychostimulants increases glutamatergic stignaling from the prefrontal cortex to the NAc. Glutamatergic signaling elevates Ca2+ levels in NAc postsynaptic elements where it activates CaMK (calcium/calmodulin protein kinases) signaling, which, in addition to phosphorylating CREB, also phosphorylates HDAC5. This results in nuclear export of HDAC5 and increased histone acetylation on its target genes (eg, NK1R[NK1 or substance P receptor]). From ref 8: Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355-367.](/cms/asset/d5a5a11f-19e1-40cb-b3cb-7126aa824e1c/tdcn_a_12130754_f0002_oc.jpg)
Cocaine-induced alterations in chromatin structure in the nucleus accumbens (NAc), the ventral portion of striatum heavily implicated as a brain reward region, have been shown to regulate behavioral responses to drugs of abuse. Pharmacological inhibition of HDACs in the NAc, which increases histone acetylation in this brain region, significantly potentiates the locomotor-activating and rewarding responses to cocaine.Citation32,Citation40,Citation41 Conversely, reducing histone acetylation by overexpressing certain HDACs, or knockdown of the HAT, CBP, results in less sensitivity to cocaine.Citation32,Citation34,Citation40 Two reports have extended these findings in rat models of cocaine self-administration, where animals are trained to press levers to receive the drug. Interestingly, delivery of the HDAC inhibitor, sodium butyrate, potentiates drug-takingCitation42 while delivery of the HDAC inhibitor, trichostatin A, attenuates it.Citation43 The explanation for these different observations is unclear, but it may involve experimental differences with the self-administration paradigm or the HDAC inhibitor used.
Cocaine alters histone acetylation through many enzymes in the NAc, but one particular HDAC, HDAC5, responds uniquely to chronic cocaine administration, raising the interesting possibility that this HDAC is involved in the behavioral transitions which occur between acute and chronic cocaine exposure (eg, drug experimentation to compulsive drug use). Chronic cocaine administration increases the phosphorylation of IIDAC5 and shuttles it out of the nucleus, permitting hyperacetylation of histones at target genes for HDACS ().Citation40 This phosphorylation reaction may be mediated by Ca2+/calmodulin-dependent protein kinase II (CaMKII), since ex vivo inhibition of CaMKII reduces the activity-induced phosphorylation of HDAC5. Consistent with its regulation by cocaine, mice deficient for HDACS display normal rewarding responses to initial cocaine exposures, but become hypersensitive when treated with a chronic course of cocaine.Citation40 Thus, pharmacological and genetic manipulations that increase histone acetylation appear to potentiate behavioral responses to cocaine and suggest that altered histone acetylation may contribute to establishment of an addicted state.
Histone H3 phosphorylation and phosphoacetylation also appear to play key roles in drug-regulated behaviors. Global levels of histone H3 phosphorylation at serine 10 are induced by acute cocaine in striatum, a process which requires the kinase MSKl.Citation19,Citation32 The function of MSK1 is behaviorally important, as mice lacking this kinase have attenuated locomotor responses to cocaine. Cocaine-induced inhibition of protein phosphatase-1 also plays an important role in IB phosphorylation in striatum (). Dopamine D1 receptor activation alters the phosphorylation of dopamine-regulated and cyclic-AMP-regulated phosphoprotein of 32kD (DARPP-32) at particular serine residues; the protein then accumulates in the nucleus to inhibit protein phosphatase-1 from dephosphorylating histone H3.Citation20 The simultaneous activation of an H3 kinase and inhibition of an H3 phosphatase results in the robust increase in H3 phosphorylation after acute cocaine exposure. The genes at which histone phosphorylation is occurring in response to cocaine remain poorly defined with an exception of c-fos, where dramatic histone phosphorylation occurs in conjunction with acetylation (phosphoacetylation).
As mentioned earlier, global histone methylation of H3K9 is also regulated by cocaine and, in turn, alters behavioral responses to the drug. For example, inhibition of a particular H3K9 histone methyltransferase, KMT1C (G9a), whose expression is regulated in the NAc by chronic cocaine administration, potentiates behavioral responses to the drug.Citation37 These findings are consistent with histone acetylation findings, since inhibition of H3K9 methylation would also be expected to enhance gene activity. Together, these data suggest that, in general, increases in gene expression potentiate behavioral sensitivity to drugs of abuse. As well, advances are being made in identifying the individual gene promoters where chronic cocaine induces alterations in H3K9 methylation and thereby regulates gene expression in the NAc.Citation37
Overall, these findings implicate changes in histone acetylation, phosphorylation, and methylation in mediating expression changes in specific sets of genes that are crucial for controlling behavioral responses to drugs of abuse.
Epigenetic mechanisms in depression
Depression is a chronic disorder characterized by many debilitating symptoms including dysphoria, anhedonia, sleep disturbances, and weight changes. Most people diagnosed with depression are prescribed some type of antidepressant medication, of which selective serotonin reuptake inhibitors (SSRIs) or mixed serotonin-norepinephrine reuptake inhibitors (SNRIs) are the most common. Unfortunately, less than 50% of patients exhibit a complete response to SSRIs, SNRIs, or related antidepressants, thus leaving a substantial portion of depressed patients with a chronic syndrome for which few effective clinical alternatives are available. Psychiatric research is thus focused on identifying new mechanisms that are involved in the pathogenesis and maintenance of depression, which may serve as novel targets for more effective therapeutics.
One of the most challenging obstacles for depression research has been the development of an animal model that accurately recapitulates human depression. While no model can effectively model all aspects of human depression (eg, suicide), some of the major symptoms such as anhedonia and sleep and weight disturbances, and their reversal by antidepressant treatment, can be studied in rodents. The pathogenesis of depressed-like states is typically modeled in rodents by chronic exposure to stress.Citation44 One such model, chronic social defeat stress, involves the repeated exposure of an experimental mouse to a series of aggressive mice over 10 days. Each day the stress begins as a brief physical encounter (typically 5 to 10 minutes) followed by a full day of sensory contact (eg, smell, sight) as the mice are separated by a screen. After 10 days of social defeat, the experimental mice develop a chronic syndrome (lasting more than a month) that is characterized by anhedonia, anxiety-like symptoms, weight loss, and loss of interest in social interaction. Importantly, SSRIs or SNRIs reverse most of these behavioral end points, making chronic social defeat stress an attractive model in which to study the molecular adaptations associated with a depressed-like state and those involved with antidepressant action.Citation45,Citation46
Brain derived neurotrophic factor (BDNF) plays a critical role in the development of the social defeat phenotype and its reversal by antidepressant treatment. It was observed that BDNF in the hippocampus is downregulated for at least 1 month after chronic social defeat stress, and that chronic antidepressant treatment reversed this downregulation.Citation46 A mechanism for this long-lasting regulation of gene expression was identified as methylation of H3K27, a repressive histone modification, that remains hypermethylated on the bdnf promoter within hippocampus for at least a month after defeat stress. While chronic antidepressant treatment of mice exposed to chronic social defeat ameliorates many of the behavioral deficits and restores bdnf mRNA to normal levels, H3K27 remains hypermethylated. The maintenance of H3K27 methyiation even after chronic antidepressant treatment suggests that BDNF expression might revert to a repressed state if drug administration were stopped. This novel epigenetic mechanism, which was proposed as a form of “molecular scar,” may describe a potential mechanism by which the symptoms of depressed patients reappear after cessation of antidepressant treatment, however, this remains speculative and further research is needed.
The recovery of bdnf expression after antidepressant treatment is likely mediated by the antidepressantinduced increase in histone H3K4 methylation and H3 polyacetylation in hippocampus, which are associated with gene activation.Citation46 Interestingly, tranylcypromine, which inhibits monoamine oxidases and is used as an antidepressant, is actually a much stronger inhibitor of the histone H3K4 demethylase KMT1A (formerly, LSD1) than it is of either monamine oxidase A or B.Citation47 Thus, it will be interesting to determine whether any of the antidepressant properties of tranylcypromine derive from its blockade of KMT1 A and the subsequent facilitation of H3K4 methylation. Arguing against this interpretation is the knowledge that several structurally unrelated monoamine oxidase inhibitors, which have not been shown to inhibit histone demethylases, are still effective antidepressants.
The increase in H3 acetylation by antidepressant treatment suggested that HDAC inhibitors may also have antidepressant-like effects. Indeed, in both the chronic social defeat model and in the forced swim test, HDAC inhibitors demonstrated antidepressant-like prosperities.Citation46,Citation48 This was especially apparent when an I ID AC inhibitor was administered in addition to an SSRI, fluoxetine. While these inhibitors target numerous HDAC5, one specific isoform, HDAC5, stood out because it was oppositely regulated by stress and antidepressant treatment.Citation46 Indeed, overexpression of HDAC5 in the hippocampus blocks the behavioral effects of chronic antidepressant treatment, suggesting that increased histone acetylation on the bdnf promoter is a key mechanism to overcome the repressive effects of H3K27 methylation. Another intriguing aspect of chronic social defeat stress is that the severity of the depression-like phenotype varies within a cohort of inbred (ie, virtually genetically identical) mice. It was observed that mice susceptible to defeat stress show significantly higher firing rates of dopaminergic neurons in the ventral tegmental area (VTA) after stress exposure compared with resilient mice. These resilient mice had normal VTA firing rates because of a stress-induced upregulation of potassium channels in this brain region. Why do certain mice upregulate protective potassium channels in the VTA while others fail to do this and become “depressed?” Perhaps an epigenetic mechanism is involved in altering the promoters of certain potassium channels to ultimately determine if the gene will be induced in response to chronic stress. If so, which life experiences trigger these chromatin remodeling events? These are important questions that may shed fundamentally new light onto an extraordinarily complex syndrome, and provide new avenues for the development of more effective antidepressants.
Another important epigenetic mechanism that may contribute to long-lasting changes in neural function and behavior is DNA methylation. Early insight into the role of DNA methylation in behavior followed from studies of maternal care that clearly demonstrate an experience-dependent rather than genetic basis for how rats treat their offspring. Rats that receive poor maternal care as pups grow up to become poor mothers to their pups. In addition to becoming poor mothers, these rats also develop long-lasting heightened anxiety and stress responses. Meaney and colleagues identified a region of the glucocorticoid receptor (GR) gene, which was hypermethylated throughout adulthood in rats who received poor maternal care. Treatment with an HDAC inhibitor not only reduced DNA methylation on the GR receptor gene but also improved anxiety and stress responses in these rats.Citation49 More recently, these studies have been translated from rats into humans by studying the hippocampus of patients who committed suicide with or without a history of child abuse. In patients with a history of child abuse, it was observed that DNA methylation on the GR gene promoter was significantly higher, while GR mRNA expression was significantly lower than patients with no history of child abuse.Citation50 While these data do not demonstrate causation, they are among the best evidence to date implicating epigenetic mechanisms in anxiety and stress and suggest that DNA methylation at the GR gene promoter (and probably other genes) in both rats and humans may contribute to this phenomenon.
Taken together, these studies demonstrate that chromatin structure is an important substrate for long-lasting changes in behavioral responses to stress and antidepressant treatments. While the precise signaling mechanisms by which environmental stresses converge on chromatin are still under investigation (eg, ), these early studies suggest the exciting possibility that pharmacological manipulation of chromatin remodeling pathways could be a novel approach to new antidepressant development.
Concluding remarks
Chromatin structure is emerging as a key substrate in the pathogenesis and maintenance of chronic psychiatric illnesses. This is important because novel therapeutics could target chromatin remodeling enzymes or chromatin itself to ultimately block or even reverse, for example, a chronically addicted or depressed state. Ultimately though, the key function of chromatin remodeling is to alter the transcription or the transcriptional potential of genes which eventually affect neural function, so any study of chromatin regulation is, in theory, inexorably linked with the study of the underlying gene activity. While extremely exciting, epigenetic research in psychiatry is still in its infancy, and far more research is needed to identify both the dysregulated genes and chromatin modifications responsible for individual psychiatric diseases. Fortunately, new advances in high- throughput sequencing are enabling such characterization of chromatin regulation and gene expression, genome-wide, at an incredible rate and resolution. Armed with these and other new research tools, epigenetic research in psychiatry is progressing at a spectacular speed, and may soon prove to be a major avenue for novel therapeutics.
Selected abbreviations and acronyms
| BDNF | = | brain derived neurotrophic factor |
| cAMP | = | cyclic adenosine monophosphate |
| CREB | = | cAMP'-response element binding protein |
| H | = | histone |
| HAT | = | histone acetyltransf erase |
| HDAC | = | histone deacetylase |
| HDM | = | histone demethylases |
| HMT | = | histone methyltransferase |
Acknowledgments: Preparation of this review was supported by grants from NIDA and NIMH, and the Medical Scientist Training Program at UT Southwestern Medical Center. Parts of this review were based on ref 8 and ref 51 with permission. The authors declare no conflicts of interest.
REFERENCES
- BirdA.Perceptions of epigenetics. Nature.200744739639817522671
- SuraniMA.HayashiK.HajkovaP.Genetic and epigenetic regulators of pluripotency.Cell. 200712874776217320511
- LugerK.RichmondTJ.The histone tails of the nucleosome. Curr Opin Genet Dev. 199881401469610403
- FelsenfeldG.GroudineM.Controlling the double helix. Nature. 200342144845312540921
- LiB.CareyM.WorkmanJL.The role of chromatin during transcription. Cell. 200712870771917320508
- KouzaridesT.Chromatin modifications and their function. Cell. 200712869370517320507
- StrahlBD.AllisCD.The language of covalent histone modifications. Nature. 2000403414510638745
- TsankovaN.RenthalW.KumarA.NestlerEJ.Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007835536717453016
- PokholokDK.HarbisonCT.LevineS.et al.Genome-wide map of nucleosome acetylation and methyiation in yeast. Cell. 200512251752716122420
- DoiM.HirayamaJ.Sassone-CorsiP.Circadian regulator CLOCK is a histone acetyltransferase. Cell. 200612549750816678094
- KawasakiH.SchiltzL.ChiuR.et al.ATF-2 has intrinsic histone acetyltransferase activity which is modulated by phosphorylation. Nature. 200040519520010821277
- ChawlaS.VanhoutteP.ArnoldFJ.HuangCL.BadingH.Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J Neurochem. 20038515115912641737
- FischleW.DequiedtF.HendzelMJ.et al.Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 20029455711804585
- LahmA.PaoliniC.PallaoroM.et al.Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A 2007104173351734017956988
- YangXJ.SetoE.The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell. Biol 2008920621818292778
- HaigisMC.GuarenteLP.Mammalian sirtuins - emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006202913292117079682
- Brami-CherrierK.LavaurJ.PagesC.ArthurJS.CabocheJ.Glutamate induces histone H3 phosphorylation but not acetylation in striatal neurons: role of mitogen- and stress-activated kinase-1 . J Neurochem. 200710169770817241117
- LiJ.GuoY.SchroederFA.et al.Dopamine D2-like antagonists induce chromatin remodeling in striatal neurons through cyclic AMP-protein kinase A and NMDA receptor signaling.J Neurochem. 2004901117113115312167
- Brami-CherrierK.ValjentE.HervéD.et al.Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J Neurosci. 200525114441145416339038
- StipanovichA.ValjentE.MatamalesM.et al.A phosphatase cascade by which rewarding stimuli control nucleosomal response. Nature. 200845387988418496528
- VakocCR.MandatSA.OlenchockBA.BlobelGA.Histone H3 lysine 9 methyiation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol Cell. 20051938139116061184
- IwaseS.LanF.BaylissP.et al.The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 20071281077108817320160
- JensenLR.AmendeM.GurokU.et al.Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am J Hum Genet. 20057622723615586325
- SuzukiMM.BirdA.DNA methyiation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008946547618463664
- ZhangX.OdomDT.KooSH.et al.Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 20051024459446415753290
- RamockiMB.ZoghbiHY.Failure of neuronal homeostasis results in common neuropsychiatrie phenotypes. Nature. 200845591291818923513
- ChahrourM.JungSY.ShawC.et al.MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 20083201224122918511691
- GollMG.KirpekarF.MaggertKA.et al.Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science. 200631139539816424344
- MillerCA.SweattJD.Covalent modification of DNA regulates memory formation. Neuron. 20075385786917359920
- OoiSK.BestorTH.The colorful history of active DNA demethylation. Cell. 20081331145114818585349
- HymanS.MalenkaR.NestlerE.Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 20062956559816776597
- KumarA.ChoiKH.RenthalW.et al.Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 20054830331416242410
- NestlerEJ.Review. Transcriptional mechanisms of addiction: role of DeltaFosB. Philos Trans R Soc Lond B Biol Sci. 20083633245325518640924
- LevineAA.GuanZ.BarcoA.et al.CREB-binding protein controls response to cocaine by acetylating histones at the fosB promoter in the mouse striatum. Proc Natl Acad Sci U S A. 2005102191861919116380431
- FischerA.SananbenesiF.WangX.DobbinM.TsaiLH.Recovery of learning and memory is associated with chromatin remodelling. Nature. 200744717818217468743
- LevensonJM.O'RiordanKJ.BrownKD.et al.Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004279405454055915273246
- MazeI.CovingtonHE.3rdLaplantQ.et al.Histone methylation in the nucleus accumbens controls behavioral responses to cocaine. Soc Neurosci Abs. In press.
- RenthalW.KumarA.XiaoG.et al.Genome wide analysis of chromatin regulation by cocaine reveals a novel role for sirtuins. Neuron. 20096233534819447090
- FreemanWM.PatelKM.BrucklacherRM.et al.Persistent alterations in mesolimbic gene expression with abstinence from cocaine self-administration. Neuropsychopharmacology. 2008331807181717851536
- RenthalW.MazeI.KrishnanV.et al.Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 20075651752917988634
- SchroederFA.PentaKL.MatevossianA.et al.Drug-induced activation of dopamine D(1) receptor signaling and inhibition of class Vll histone deacetylase induce chromatin remodeling in reward circuitry and modulate cocaine-related behaviors. Neuropsychopharmacology. 2008332981299218288092
- SunJ.WangL.JiangB.et al.The effects of sodium butyrate, an inhibitor of histone deacetylase, on the cocaine- and sucrose-maintained self-administration in rats. Neurosci Lett. 2008441727618599214
- RomieuP.HostL.GobailleS.et al.Histone deacetylase inhibitors decrease cocaine but not sucrose self-administration in rats. J Neurosci. 2008289342934818799668
- KrishnanV.NestlerEJ.The molecular neurobiology of depression. Nature. 200845589490218923511
- BertonO.McClungCA.DileoneRJ.et al.Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 200631186486816469931
- TsankovaN.BertonO.RenthalW.et al.Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006951952516501568
- LeeMG.WynderC.SchmidtDM.McCaffertyDG.ShiekhattarR.Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol. 20061356356716793513
- SchroederFA.LinCL.CrusioWE.AkbarianS.Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 200762556416945350
- WeaverIC.CervoniN.ChampagneFA.et al.Epigenetic programming by maternal behavior. Nat Neurosci. 2004784785415220929
- McGowanPO.SasakiA.D'AlessioAC.et al.Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 20091234234819234457
- RenthalW.NestlerEJ.Epigenetic mechanisms in drug addiction. Trends Mol Med. 20081434135018635399