
Abstract
The contributions of mesenchymal stem cells (MSCs) to tumour growth and stroma formation are poorly understood. Tumour cells can transfer genetic information and modulate cell signalling in other cells through the release of extracellular vesicles (EVs). We examined the contribution of EV-mediated inter-cellular signalling between bone marrow MSCs and tumour cells in human cholangiocarcinoma, highly desmoplastic cancers that are characterized by tumour cells closely intertwined within a dense fibrous stroma. Exposure of MSCs to tumour cell–derived EVs enhanced MSC migratory capability and expression of alpha-smooth muscle actin mRNA, in addition to mRNA expression and release of CXCL-1, CCL2 and IL-6. Conditioned media from MSCs exposed to tumour cell–derived EVs increased STAT-3 phosphorylation and proliferation in tumour cells. These effects were completely blocked by anti-IL-6R antibody. In conclusion, tumour cell–derived EVs can contribute to the generation of tumour stroma through fibroblastic differentiation of MSCs, and can also selectively modulate the cellular release of soluble factors such as IL-6 by MSCs that can, in turn, alter tumour cell proliferation. Thus, malignant cells can “educate” MSCs to induce local microenvironmental changes that enhance tumour cell growth.
Bone marrow–derived mesenchymal stem cells (MSCs) are a potential source of tissue replacement because of their regenerative ability and multipotent capability. Under the appropriate environment, these cells can be induced to differentiate into osteocytes, adipocytes, chondrocytes and myocytes (Citation1–Citation3). Understanding the contributions of MSCs to tumour biology is of importance because they may result in new therapeutic or preventive paradigms. Within the tumour microenvironment, MSCs can differentiate into myofibroblasts, cancer-associated fibroblasts, fibrocytes or pericytes and thereby represent a potential source of tumour stroma and desmoplasia (Citation4–Citation6). A contribution of interactions between MSCs within tumour stroma and cancer cells to tumour progression and metastases has been identified (Citation7–Citation9). MSCs may contribute to tumour propagation or dissemination by preventing recognition of the tumour cells by the immune system or by promoting tumour cell invasiveness (Citation10, Citation11). However, MSCs could also suppress tumour growth (Citation12–Citation15). Thus, while MSCs may interact with tumour cells, the consequences of these interactions and impact on tumour behaviour warrant definition, and likely depend on other factors.
Amongst the most highly desmoplastic tumours are cholangiocarcinomas, tumours arising from the biliary tract. These tumours are characterized by tumour cells closely intertwined with a dense fibrous stroma (Citation16–Citation19). Although this stromal desmoplastic reaction has long been recognized as a hallmark histological feature, the contribution of the mesenchymal compartment and desmoplastic stroma to tumour formation and progression has only recently been recognized. A crucial role for cancer-associated fibroblasts and activated macrophages in these cancers is emerging (Citation17, Citation18) (Citation20). Despite this recent interest, the cellular origins and mechanistic contribution of tumour stroma to tumour growth remain poorly understood. In particular, the source of tumour stroma and the nature of the interactions between tumour cells and stroma are unknown. Tumour cells can interact with other cellular elements within the local microenvironment by cell–cell interactions and paracrine mechanisms through the production and release of a variety of growth factors, chemokines and matrix-degrading enzymes that can enhance the proliferation and invasion of tumour (Citation21). An alternative mechanism by which tumour cells can interact with the local microenvironment involves inter-cellular communication involving the release of extracellular vesicles (EVs) such as exosomes (Citation22). These EVs can be released from normal as well as tumour cells (Citation23–Citation26), and have been shown to contain proteins and RNAs such as non-coding RNAs (Citation26, Citation27). We have recently shown that tumour cells can transfer genetic information by release of EVs that can modulate recipient cell behaviour (Citation25). Thus, our aims were to examine the effects of tumour cell–MSC interactions involving EVs and their contribution to tumour stroma formation and tumour growth.
Materials and methods
Cell lines and culture
For these studies, we used KMBC and HuCCT1 human cholangiocarcinoma cells and H69 human non-malignant cholangiocyte cells. KMBC cells were cultured in Dulbecco's modified Eagle's medium (DMEM) high-glucose medium (HyClone, Logan, UT), containing 10% foetal bovine serum (FBS) and 1% antibiotic–antimycotic (Life Technologies, Grand Island, NY). HuCCT-1 cells were cultured in CMRL 1066 media with 10% FBS, 1% L-glutamine and 1% antibiotic–antimycotic as previously described (Citation28). H69 cells were cultured in hormonally supplemented medium, composed of DMEM/nutrient mixture F-12 Ham (GIBCO BRL, Gaithersburg, MD) (3:1) containing adenine, insulin, triiodothyronine-transferrin, hydrocortisone, epinephrine, epidermal growth factor, penicillin/streptomycin and 10% FBS. The human bone marrow vascular stromal fraction was isolated from a healthy donor using Histopaque-1077 (Sigma-Aldrich, St. Louis, MO) following density gradient protocols. Bone marrow–derived MSCs were generated following culture in alpha Minimum Essential medium (αMEM) (Invitrogen, Carlsbad, CA) containing 16.5% FBS according to a previously reported method (Citation1, Citation29) (Citation30). For all studies, vesicle-depleted medium was prepared by ultracentrifugation of cell-culture medium at 100,000 g for 70 minutes (Beckman coulter, Optima L- 80XP) to spin down any pre-existing vesicular content.
Isolation and characterization of EVs
Isolation of EVs was performed as we have described (Citation25, Citation31). Briefly, cells (1×106) were plated in 11 ml of vesicle-depleted medium on collagen coated 10-cm plates. After 3 days, the medium was collected and sequential centrifugation was performed. The medium was first centrifuged at 300 g for 10 minutes, and then at 2,000 g for 20 minutes in 4°C to remove cells. The supernatant was then centrifuged at 10,000 g for 70 minutes at 4°C. The supernatant was further ultracentrifuged at 100,000 g for 70 minutes at 4°C to pellet EVs, which were then washed by suspending in phosphate-buffered saline (PBS) and ultracentrifuged at 100,000 g for 70 minutes at 4°C. The final pellet was resuspended with 200–500 µL of PBS and stored at −80°C. The protein yield was measured using a bicinchoninic acid protein assay kit (Thermo Fisher, Rockford, IL). The number of EVs was quantitated using NanoSight LM10 (Malvern Instruments, Malvern, UK). Electron microscopy was performed on whole mounts of isolated EVs using 2.5% glutaraldehyde, postfixed with 1% osmium tetroxide and embedded and stained with uranyl acetate and lead citrate (or 4% paraformaldehyde and negatively stained with 1% neutral aqueous uranyl acetate). Transmission electron microscopy was performed using an EM208S (Philips, Eindhoven, The Netherlands).
Quantitative real-time PCR (qRT-PCR)
RNA was treated with RNase-free DNase I (Qiagen, Valencia, CA). One microgram of RNA was reverse-transcribed to cDNA using iScript cDNA Synthesis Kit (BioRad, Hercules, CA), and qRT-PCR was performed using a LightCycler 96 System (Roche Diagnostics, Mannheim, Germany) with SYBR green I (SYBR® Advantage® qPCR Premix, Clontech, Mountain View, CA) as the detection fluorophore and the following PCR primers: Fibroblast activation protein (FAP): forward: 5′-ATCTATGACCTTAGCAATGGAGAATTTGT-3; reverse: 5′-GTTTTGATAGACATATGCTAATTTACTCCCAAC-3; Vimentin: forward: 5′-CCTTGAACGCAAAGTGGAATCT-3; reverse: 5′-CCACATCGATTTGGACATGCT-3; Alpha-smooth muscle actin (α-SMA): forward: 5′-GCAGCCCAGCCAAGCACTGT-3; reverse: 5′-TGGGAGCATCGTCCCCAGCA-3; IL-6: forward: 5′-CACCCCTGACCCAACCACAAAT- 3; reverse: 5′-TCCTTAAAGCTGCGCAGAATGAGA-3; CXCL1/GRO-α: forward: 5′- CTCTACCTGCACACTGTCCTATTA-3′, reverse: 5′-ATCCGCCAGCCTCTATCA-3; CCL2/MCP-1: forward: 5′- GTCTCTGCCGCCCTTCTGTG-3′, reverse: 5′- AGGTGACTGGGGCATTGATTG-3′.
Immunoblot analysis
Cells were lysed using cOmplete Lysis-M, EDTA-free and cOmplete Mini, EDTA-free, Protease Inhibitor Cocktail Tablet and PhosSTOP Phosphatase inhibitor (Roche, Indianapolis, IN). Equivalent amounts of protein lysates were mixed with 4X LDS Sample Buffer (Life Technologies, Grand Island, NY), separated on 4–12% Bis-Tris Gels (Life Technologies, Grand Island, NY) and then transferred to nitrocellulose membranes, blocked with Odyssey Blocking Buffer (LI-COR, Lincoln, NE) for 30 minutes and incubated overnight at 4°C with the respective primary antibodies: mouse monoclonal antibody anti-α-SMA (1:1,000) (Sigma-Aldrich, St. Louis, MO), mouse monoclonal antibody anti-STAT3(124H6) (1:1,000) (Cell Signaling, Beverly, MA), rabbit monoclonal antibody anti-phospho (Y705)-STAT3 (1:500) (Abcam, Cambridge, UK) and goat polyclonal anti-Actin (1:5,000) (Santa Cruz, Dallas, TX). The membrane was washed thrice for 10 minutes with TBS-T (25 mM Tris-HCl, pH 7.4, 125 mm NaCl, 0.05% Tween 20) and then incubated with Alexa Fluor 680 anti-mouse (1:5,000), anti-rabbit (1:5,000) or anti-goat IgG (1:5,000) (Life Technologies, Grand Island, NY) for 20 minutes. Proteins were visualized and quantitated using the Odyssey imaging System (LI-COR, Lincoln, NE). Relative expression was determined by probing the same membrane against Actin.
Immunocytochemistry
MSCs were plated in 6-well plates and cultured for 28 days. KMBC-EVs (2×1010 particles/well) and H69-EVs (2×1010 particles/well) were added to the cell culture every 7 days. After 28 days, cells were collected, plated on coverslips, fixed using 4% paraformaldehyde, permeabilized and incubated with anti-α-SMA antibody (Abcam, Cambridge, MA), rabbit polyclonal antibody anti-FAP (10 µg/ml) (Abcam, Cambridge, UK) or goat polyclonal antibody anti-Vimentin (1:50) (Santa Cruz, Dallas, TX), overnight at 4°C. Cells were washed and incubated with secondary antibody for 1 hour, then mounted with Prolong Gold Antifade Reagent with DAPI (Life Technologies, Grand Island, NY) and visualized by fluorescence microscopy (Olympus IX71, Olympus America, Center Valley, PA).
Preparation of MSC-conditioned medium
MSCs (1×104/well) were cultured with vesicle-depleted medium in the presence or absence of KMBC-EVs (1×1010 particles/ml) on 6-well plates for up to 48 hours. The conditioned medium (CM) was harvested by filtering culture supernatant through a 0.20-micron Nalgene filter unit (Thermo scientific, Rockford, IL).
Cell proliferation assay
KMBC cells were plated (1×104/well) into 96-well collagen coated plates and cultured. At selected times, proliferation was assessed using MTS solution (Promega, Madison, WI) and a FLUOstar Omega Microplate Reader (BMG Labtech, Cary, NC). Background correction was performed by subtracting background fluorescence from wells without cells. For antibody inhibition studies, KMBC cells were cultured in the presence of stem cell CM containing 5 µg/ml of normal mouse IgG, mouse monoclonal anti- CXCL1 or mouse monoclonal anti-IL-6R antibody (R&D Systems, Minneapolis, MN), for 72 hours.
Migration assay
For MSC studies, 8×104 cells were suspended in 200 µl serum-free medium in the presence or absence of 6.0×109 KMBC-EVs/well and loaded onto the upper compartment of Transwell (Corning, Lowell, MA) 24-well plates with a pore size of 8.0 µm. Moreover, 8×104 MSCs on the upper compartment were cultured with CM from KMBC treated with or without GW4869, an inhibitor of ceramide synthesis that reduces cellular release of EVs (Calbiochem, Darmstadt, Germany) for 24 hours. For KMBC studies, 5.0×104 cells were loaded onto the upper compartment. Serum-free medium (500 µl) was added to the bottom. After 12 hours, cells that had migrated through the membrane were fixed and stained using Diff-Quik (Astral Diagnostics, West Deptford, NJ). Migrated cells were identified and quantitated using a microscope and average counts from 5 or more fields of cells were obtained for each group.
Cytokine and chemokine profiling
Culture supernatant was obtained from MSCs incubated in the presence or absence of KBMC-EVs for 72 hours, and expression of released proteins was measured using the Human 64-Plex Cytokine/Chemokine Panel and Human MMP & TIMP Panels (Eve Technologies, Calgary, AB, Canada). Analysis was performed on 3 or more independent samples for each condition.
Statistical analysis
Data were expressed as the mean and standard error from at least 3 replicates. Comparisons between groups were performed using the 2-tailed Student's t-test, and results were considered to be statistically significant when p<0.05.
Results
Characterization of tumour cell–derived EVs
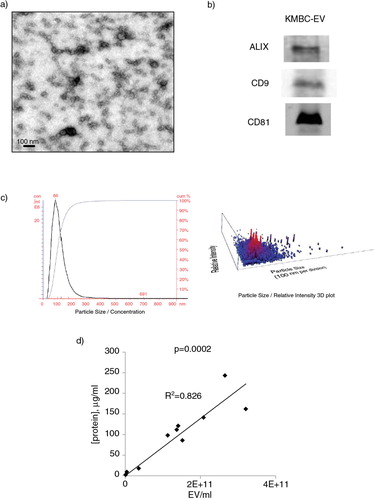
We first characterized the EVs isolated from KMBC tumours in culture. Electron microscopy showed that the isolated vesicles were spherical (a), and density gradient ultracentrifugation revealed a mean density of 1.12 g/ml. Isolated vesicles expressed ALIX, CD9 and CD81 (b). Nanoparticle tracking analysis identified a peak size vesicle diameter of 86 nm (c). Thus, a homogeneous population of EVs with features consistent with exosomes can be obtained from cholangiocarcinoma cells in culture. The term “exosomes” refers to a specific subset of EVs with a distinctive biogenesis which was not directly ascertained, and thus we refer to isolated vesicles as EVs. A highly positive correlation was observed between EV protein concentration and quantity derived from nanoparticle tracking analysis with a correlation coefficient, r=0.91, and p=0.0002 (d).
Fig. 1. Characterization of tumour cell–derived extracellular vesicles (EVs). (a) Transmission electron microscopy was performed on a whole mount of particles isolated from KMBC cells using an ultracentrifugation method. (b) Western blot analysis for proteins associated with exosomes in isolated KMBC-EVs. (c) Quantitation using Nanosight nanoparticle tracking analysis. KMBC cells produced EVs in cell culture with a mean size of 137±60 nm and a peak size of 86 nm. A homogenous population with size and morphology consistent with that of exosomes was obtained. (d) Protein concentration was determined using BCA assay of EVs isolated from KMBC cells.
Tumour cell–derived EVs induce fibroblastic differentiation in human bone marrow MSCs
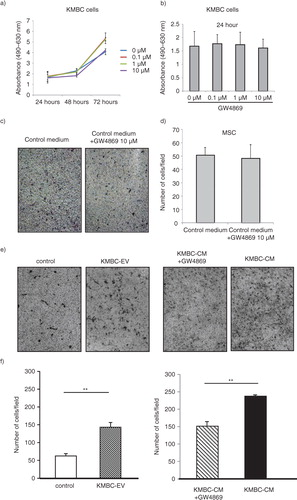
Several potential sources of tumour stroma have been postulated, such as expansion of a resident fibroblast population, or differentiation of a pluripotent stem population that is either resident within tissues, or has been recruited. We hypothesized that tumour cell–derived EVs might contribute to tumour stroma formation by inducing the differentiation of bone marrow–derived MSCs that are recruited to the tissues. We therefore examined the effect of exposure to tumour cell–derived EVs on fibroblastic differentiation of human bone marrow MSCs. MSCs were cultured in medium containing 1×1010 particles/ml KMBC or HuCCT1-derived EVs on 6-well plates and the expression of myofibroblastic/fibroblastic markers such as α-SMA, vimentin or FAP were examined. MSCs in culture adopted morphological changes such as elongated cellular processes which were enhanced by exposure of MSCs to KMBC-EVs. The expression of α-SMA and FAP mRNA was significantly increased in MSCs incubated with either KMBC-EVs or HuCCT1-EVs compared to controls incubated in medium without EVs after 48 hours (a). Moreover, we cultured MSCs with KMBC-conditioned medium (CM) and KMBC-CM deprived of EVs under the same conditions. KMBC-CM increased the most expression of α-SMA and FAP mRNA on MSCs. KMBC-CM deprived of EVs significantly decreased these expressions on MSCs compared with KMBC-CM and significantly increased these expressions compared with control (b). Quantitative immunoblot analysis identified an increase in the expression of α-SMA, FAP and vimentin protein in MSCs incubated with KMBC-EVs for 14 days (a). Similar observations were made on immunofluorescence microscopy for expression in long-term 28-day cultures of MSCs with KMBC-EVs; however, an increase in vimentin was not detected (b). In contrast, EVs derived from non-malignant H69 cells did not increase α-SMA expression after 28 days. These studies suggest that exposure to tumour cell–derived EVs can enhance fibroblastic differentiation of MSCs. To examine the effect of tumour cell–derived EVs on stem cell behaviour, we next studied whether KMBC cell–derived EVs could modulate the migratory ability of MSCs, using transwell cell migration assays. Incubation of MSCs with KMBC-EVs resulted in increased cell migration after 12 hours compared to control MSCs that were not exposed to KMBC-EVs (). Moreover, migration of MSCs cultured with CM from KMBC pre-incubated with GW4869 was decreased compared to migration of MSCs cultured with CM from KMBC. GW4869 did not affect KMBC cell viability (a, b) or migration of MSCs. These findings support the presence of interactions whereby malignant transformed cells can modulate cellular behaviour within the local microenvironment through release of EVs that can manipulate genetic and phenotypic changes in stem cells.
Fig. 2. Tumour cell–derived extracellular vesicles (EVs) induce the expression of myofibroblast markers in hBM-MSCs. (a) MSCs were cultured in the presence or absence of EVs derived from KMBC or HuCCT-1 cholangiocarcinoma cells. Vimentin, FAP and α-SMA mRNA expression was assessed by qRT-PCR. The expression of FAP and α-SMA was significantly increased in MSCs incubated with tumour cell–derived EVs. Bars express the mean value±SEM of 3 separate studies. *p<0.05. (b) MSCs were cultured in the presence or absence of KMBC-EVs, conditioned medium from KMBC (CM), or condition medium deprived of EVs (EVD-CM). Vimentin, FAP and α-SMA mRNA expression were assessed by qRT-PCR. FAP and α-SMA expression was increased by CM, which was decreased by EVD-CM. Bars express the mean value±SEM of 3 separate studies. *p<0.05.

Fig. 3. Tumour cell–derived extracellular vesicles (EVs) induce the protein expression of myofibroblast markers in hBM-MSCs. (a) Western blotting analysis of α-SMA, FAP and vimentin protein expression in MSCs. The expression of α-SMA, FAP and vimentin was increased in MSCs incubated with KMBC-EVs for 14 days. Bars represent quantitative densitometric data showing the mean value±SEM of 3 separate studies. *p<0.05, *p<0.01. (b) Immunofluorescence microscopy of α-SMA expression in MSCs after 28-day culture. MSCs were cultured in the presence or absence of EVs derived from malignant KMBC cells or non-malignant H69 cells. An increase in α-SMA is seen in MSCs cultured in the presence of KMBC but not H69-derived EVs.
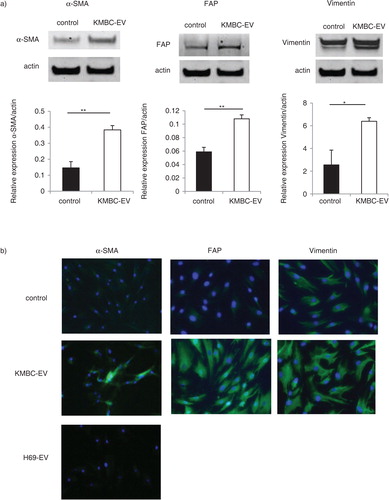
Fig. 4. KMBC-derived extracellular vesicles (EVs) increase migration of MSCs. (a, b) KMBC cells were incubated with varying concentrations of GW4869, an inhibitor of ceramide synthesis that reduces EV release. Cell viability was assessed using an MTS assay. Bars express the mean value±SEM of 3 separate studies. (c–f) Migration of MSCs across a basement membrane was assessed using transwell migration assays. Representative staining of migratory cells and quantitative data representing the mean±SEM of 3 separate studies are shown. (c, d) The effect of control medium with or without GW4869 on migration of MSCs was assessed. GW4869 did not significantly affect migration of MSCs. (e, f) MSCs were cultured alone or in the presence of KMBC cell–derived EVs (KMBC-EVs), conditioned medium (CM) from KMBC cells or CM from KMBC cells incubated with GW4869. KMBC-EVs significantly increased migration of MSCs. Similarly, exposure to CM from KMBC cells also increased migration, but this was reduced in the presence of GW4869.
Cytokine and chemokine release by human bone marrow MSCs exposed to tumour cell–derived EVs
Release of bioactive secreted factors such as cytokines and chemokines is an established mechanism of inter-cellular communication and has been extensively characterized during tumour growth and progression. In order to understand the effects of tumour cell–derived EVs on the local microenvironment, we examined the effects of exposure to KMBC-EVs on the release of biologically active proteins by MSCs. Culture supernatant was obtained from MSCs cultured in the presence or absence of KMBC-EVs for 72 hours, and protein analysis for selected cytokines, chemokines, metalloproteases and their inhibitors was performed. The concentration of each protein was also determined in isolated KMBC-EV preparations to exclude the potential for cross-over of tumour cell–released factors. Most of the proteins that were analyzed were not detected in CM obtained from MSCs in culture (Table ). However, several proteins were identified that were not only released by MSCs, but whose concentrations in was further increased in culture supernatant from MSCs incubated with KMBC-EVs. These included chemokine (C-X-C motif) ligand 1 (CXCL1/GRO-α), chemokine (C-C motif) ligand 2 (CCL2/MCP-1), chemokine (C-X3-C motif) ligand 1 (CX3CL1/Fractalkine), platelet-derived growth factor and interleukin-6 (IL-6), which were increased by 1.2-to-10.2-fold in the presence of KMBC-EVs compared to controls (Table , a). Furthermore, the expression of IL-6, CXCL1 and CCL2 mRNA was also significantly increased in MSCs treated with KMBC-EVs by quantitative RT-PCR analyses compared to control (b), although we did not detect an increase in CX3CL1 mRNA in MSCs treated with KMBC-EVs (data not shown). These results indicate that MSCs treated with KMBC-EVs secrete these cytokines and chemokines. The expression of IL-6 and CCL-2 mRNA was increased, whereas the expression of CXCL1 mRNA was decreased in KMBC-EVs compared to KMBC cells (c). These observations suggest that mRNA in EVs could potentially affect expression of the corresponding proteins directly after uptake of EVs by MSCs.
Fig. 5. KMBC cell–derived extracellular vesicles (EVs) modulate cytokine and chemokine release by MSCs. (a) MSCs were cultured in the presence or absence (controls) of KMBC-derived EVs. Conditioned medium was obtained and cytokine and chemokine concentrations determined. Exposure to KMBC-EVs increased CXCL1, CCL2, CX3CL1, PDGF and IL-6 release by MSCs. (b) IL-6, CXCL1 and CCL2 mRNA expression was quantitated by qRT-PCR in MSCs exposed to KMBC-EVs compared to MSCs without KMBC-EVs (controls). The expression of IL-6 mRNA (p=0.021), CXCL1 mRNA (p=0.039) and CCL2 mRNA (p=0.030) were significantly increased in MSCs treated with KMBC-EVs compared to controls. (c) IL-6, CXCL1 and CCL2 mRNA expression was quantitated by qRT-PCR in KMBC cells and KMBC-EVs. Compared to KMBC cells, IL-6 and CCL2 mRNA expression were significantly increased whereas the expression of CXCL1 mRNA was decreased in KMBC-EVs. Bars express the mean value±SEM of 3 separate studies.*p<0.05, **p<0.01, ***p<0.001.
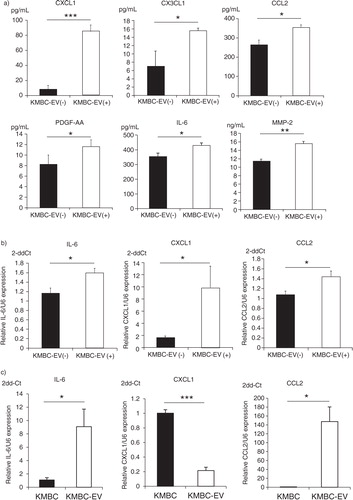
Table I. Selective changes in secretome of bone marrow–derived mesenchymal stem cells (MSCs)
KMBC proliferation and migration in response to MSCs exposed to tumour cell–derived EVs
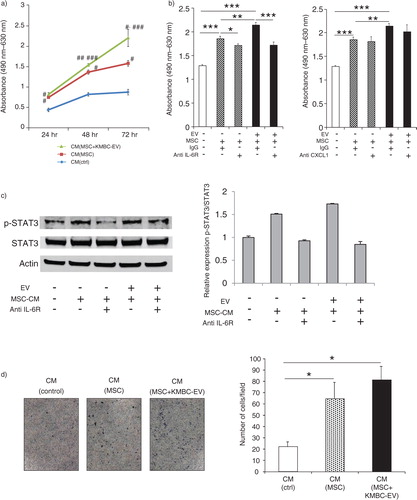
IL-6 is mitogenic for malignant cholangiocytes. In previous studies, we have examined the role of autocrine or paracrine signalling involving tumour cell–derived IL-6 in cholangiocarcinoma growth (Citation32–Citation34). The tumour stroma and non-tumour cells can also represent sources of growth factors such as IL-6. Our observations support the release of IL-6 from MSCs in response to EV-mediated interactions between tumour cells and MSCs. To examine the biological effects of MSC-secreted IL-6, we examined the effect of MSC CM on tumour cell proliferation. KMBC cells were cultured with CM obtained from MSCs incubated with KMBC-EVs. CM from MSCs cultured in the absence of KMBC-EVs or control media were used as controls. Tumour cell proliferation was assessed for up to 72 hours. We observed that the proliferation of KMBC cells was significantly increased in the presence of CM from MSCs that had been exposed to KMBC-EVs, compared to controls (a). Furthermore, proliferation of KMBC cells incubated with CM from MSCs or from MSCs exposed to KMBC-EVs was significantly blocked with anti-IL-6R antibody, although anti-CXCL-1 did not significantly alter KMBC proliferation (b). These results indicate that IL-6 secreted by MSCs treated with KMBC-EVs increase KMBC cells proliferation. Moreover, anti-IL-6R antibody blocked enhanced STAT3 activation in these cells in response to CM from either MSCs or MSCs exposed to KMBC-EVs (c). We then examined the effect of stem cell–derived factors on KMBC migration. KMBC migration was evaluated using transwell migratory assays in response to incubation with CM from MSCs, MSCs exposed to KMBC-EVs or cell-free media (control) (d). Cell migration was assessed after 12 hours. Compared to control media (without cells), KMBC migration was increased by CM from MSCs, although no difference was apparent between CM obtained in the presence or absence of KMBC-EVs (d).
Fig. 6. Conditioned medium from mesenchymal stem cells (MSCs) exposed to tumour cell–derived extracellular vesicles (EVs) enhances tumour cell proliferation. (a) KMBC cells were incubated with conditioned medium obtained from MSCs cultured in the presence or absence of KMBC-EVs. Cell proliferation was assessed using an MTS assay. Compared to controls, conditioned medium (CM) from MSCs exposed to KMBC-EVs increased KMBC cell proliferation. *p<0.001 vs. CM (control), **p<0.0001 vs. CM (control), ***p<0.05 vs. CM (MSCs). (b) Cell proliferation was assessed after 72 hours using MTS assay. Anti-IL-6R antibody, but not anti-CXCL-1 antibody, blocked enhanced KMBC cell proliferation in response to conditioned medium from MSCs exposed to tumour-derived EVs. Bars express the mean value±SEM of 3 separate studies. *p<0.05, **p<0.01, ***p<0.001. (c) Immunoblot analyses of p-STAT3/STAT3 protein expression in KMBC cells. Anti-IL-6R antibody blocked enhanced STAT3 activation in response to CM from MSCs exposed to tumour-derived EVs. Bars represent quantitative densitometric data from 3 replicates. (d) KMBC cell migration was assessed using a transwell migration assay. KMBC cells were treated with CM from MSCs and MSCs exposed to KMBC-EVs. Representative images of migrated cells and quantitative data representing the mean (±SEM) number of migrated cells from 3 separate studies. Tumour cell migration was increased by MSC-CM but was not further enhanced by prior exposure of MSCs to KMBC-EVs.
Discussion
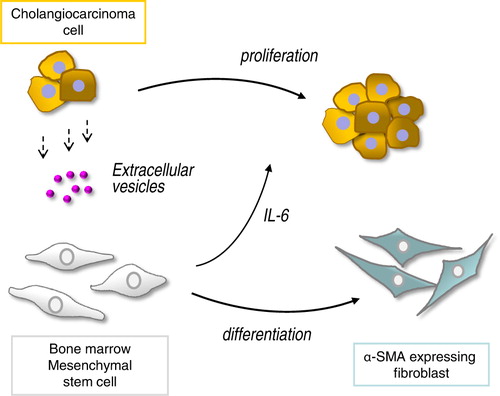
These studies show that EV transfer from malignant cholangiocytes can increase fibroblast-like activity by MSCs. Furthermore, exposure of MSCs to tumour cell–derived EVs results in a selective alteration of mRNA expression and release of cytokines/chemokines such as IL-6 that can, in turn, alter tumour cell growth. Thus, tumour cells can “educate” MSCs to modulate the microenvironment and thereby facilitate tumour growth (). This is not previously described and is a unique mechanism by which cholangiocarcinoma cells could modulate the microenvironment and facilitate tumour growth. These findings offer new insights into tumour stromal interactions, and their contribution to cholangiocarcinoma growth.
Fig. 7. Malignant cells “educate” mesenchymal stem cells (MSCs) to promote tumour growth through extracellular vesicles. Tumour cells release extracellular vesicles such as exosomes that can be taken up by bone marrow–derived MSCs. This results in selective modulation of mRNA expression and release of cytokines/chemokines such as IL-6 from MSCs, as well as in phenotypic changes consistent with fibroblast-like activity. The altered secretome and fibroblastic differentiation contribute to both tumour cell growth and stromal development in cholangiocarcinoma.
The presence of chronic biliary tract injury and inflammation predisposes to cholangiocarcinoma (Citation35). Bone marrow MSCs can be recruited to sites of inflammation (Citation36). Their subsequent contribution is undefined, with the current paradigm being that they are involved in tissue repair. Our findings indicate that the presence of these stem cells in the local microenvironment can result in their recruitment and education to enhance cell growth, as well as their fibroblastic differentiation that may eventually contribute to the desmoplastic stroma. Defining the mechanisms by which stem cells are recruited to the biliary tract during chronic inflammation may lead to strategies to prevent progression of biliary tract scarring, and tumour progression. Such preventive strategies may be particularly appropriate for conditions such as hepatolithiasis, fluke infection or primary sclerosing cholangitis, all of which are associated with a predisposition to cholangiocarcinoma.
Recent studies have indicated that MSCs can also exhibit a marked tropism for tumours. Indeed, the ability of MSCs to migrate to primary tumours may be exploited to use MSCs as candidate vehicles to deliver anti-tumour agents (Citation37). However, there is some controversy regarding the mechanisms by which these cells exert effects on tumour progression. Cell-derived EVs such as exosomes can carry lipids, proteins, mRNAs and microRNAs that can be transferred to a recipient cell following fusion of the exosome with the target cell membrane and internalization of its content (Citation38). Similar to our observations, tumour cell–derived exosomes from other malignancies such as breast, ovarian and gastric cancers can enhance myofibroblast-like differentiation of MSCs (Citation39–Citation41). However, the contribution of MSCs that have been exposed to tumour cells on subsequent tumour growth is unclear. Some, but not all, studies indicate that tumour growth or metastases may be enhanced by co-injection of MSCs with tumour-exosomes (Citation7, Citation42) (Citation43, Citation44) (Citation45) . A recent study showed that human umbilical cord MSCs could suppress cholangiocarcinoma growth (Citation46). However, the effects of MSCs that are present in the presence of tumours, and whose behaviour may have been modulated by tumour cell–MSC interactions have not been defined. Discrepancies may also result from differences in tumour models studied, heterogeneity of MSCs or other undefined environmental factors (Citation47), and emphasize the need for detailed investigation of tumour cell–EV interactions.
The cellular origins of tumour stroma in cholangiocarcinoma are not well characterized, and multiple sources are possible. In mouse models of inflammation-induced gastric cancer, more than a fifth of cancer-associated fibroblasts may originate from bone marrow–derived MSCs (Citation5). Cholangiocarcinoma-derived EVs increased the expression of myofibroblast markers such as α-SMA and FAP and enhanced migratory capability of human bone marrow MSCs, suggesting that tumour EVs could induces a phenotypic switch from MSCs to myofibroblast-like cells within the tumour stroma. Such a mechanism could enhance stem cell migration to the local tumour microenvironment, differentiation and tumour stroma formation, and eventually contribute to the growth of cancer.
The release of biologically active mediators such as IL-6 in response to tumour cell–derived EVs is highly relevant to tumour growth. The contribution of IL-6 to cholangiocarcinoma growth is recognized (Citation32, Citation33) (Citation34, Citation48) (Citation49) , and active modulation of environmental IL-6 by tumour cells provides a plausible mechanism for tumour growth. These observations may be important in other conditions that are characterized by aberrant IL-6 release, such as tissue injury, or other cancers. Indeed, STAT3 activation by IL-6 from MSCs has been reported to promote the proliferation and metastasis of osteosarcoma (Citation50). Our study also indicated that STAT3 activation by IL-6 from MSCs increases proliferation of cholangiocarcinoma. Furthermore, IL-6 released from MSCs can increase the tumour cell secretion of endothelin-1 which can induce the activation of Akt and ERK in endothelial cells, and enhance their recruitment to tumour and angiogenesis (Citation51). Although other factors such as CXCL1 that were also released from MSCs in response to tumour-derived EVs did not alter cell proliferation, we speculate that these factors could have other effects that could modulate tumour growth or spread. CXCL1 plays a role in the processes of angiogenesis, inflammation, wound healing and tumorigenesis (Citation52, Citation53). CXCL1 secreted from MSCs may participate in these processes. The presence of IL-6, CXCL1 and CCL2 mRNA within KMBC-EVs also raise the possibility that EV-enclosed mRNA could directly affect protein expression within MSCs. Similarly, we have identified the presence of EVs from MSCs in culture, and the potential exists for MSCs to direct tumour cell responses through release of EVs. Further study to examine the roles of individual MSC-derived factors released from “educated” stem cells, as well as their combinatorial effects, is warranted.
Conclusions
Our studies raise several new insights into the involvement of EV-mediated interactions between tumour and MSCs, and the contribution of these interactions to tumour growth through stromal formation and modulation of cell proliferation. The mechanisms whereby tumour cells can educate stem cells to modulate the microenvironment to facilitate tumour growth could be targeted therapeutically, and offer new opportunities for preventive or therapeutic intervention for cancers.
Conflict of interest and funding
The authors have not received any funding or benefits from industry or sources other than those listed in the acknowledgements to conduct this study.
Acknowledgements
Financial support was provided in part by Grants DK069370 and TR000884 from the National Institutes of Health and the Mayo Clinic Center for Regenerative Medicine.
References
- Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999; 284: 143–7.
- Liechty KW, MacKenzie TC, Shaaban AF, Radu A, Moseley AM, Deans R et al. Human mesenchymal stem cells engraft and demonstrate site-specific differentiation after in utero transplantation in sheep. Nat Med. 2000; 6: 1282–6.
- De Bruyn C, Najar M, Raicevic G, Meuleman N, Pieters K, Stamatopoulos B et al. A rapid, simple, and reproducible method for the isolation of mesenchymal stromal cells from Wharton's jelly without enzymatic treatment. Stem Cells Dev. 2011; 20: 547–57.
- Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One. 2009; 4: e4992.
- Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011; 19: 257–72.
- Ogawa M, LaRue AC, Drake CJ. Hematopoietic origin of fibroblasts/myofibroblasts: its pathophysiologic implications. Blood. 2006; 108: 2893–6.
- Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007; 449: 557–63.
- Lu YR, Yuan Y, Wang XJ, Wei LL, Chen YN, Cong C et al. The growth inhibitory effect of mesenchymal stem cells on tumor cells in vitro and in vivo. Cancer Biol Ther. 2008; 7: 245–51.
- Hamada H, Kobune M, Nakamura K, Kawano Y, Kato K, Honmou O et al. Mesenchymal stem cells (MSC) as therapeutic cytoreagents for gene therapy. Cancer Sci. 2005; 96: 149–56.
- Ohno S, Tachibana M, Fujii T, Ueda S, Kubota H, Nagasue N. Role of stromal collagen in immunomodulation and prognosis of advanced gastric carcinoma. Int J Cancer. 2002; 97: 770–4.
- Keleg S, Buchler P, Ludwig R, Buchler MW, Friess H. Invasion and metastasis in pancreatic cancer. Mol Cancer. 2003; 2: 14.
- Qiao L, Xu Z, Zhao T, Zhao Z, Shi M, Zhao RC et al. Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res. 2008; 18: 500–7.
- Li L, Tian H, Chen Z, Yue W, Li S, Li W. Inhibition of lung cancer cell proliferation mediated by human mesenchymal stem cells. Acta Biochim Biophys Sin (Shanghai). 2011; 43: 143–8.
- Khakoo AY, Pati S, Anderson SA, Reid W, Elshal MF, Rovira II et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi's sarcoma. J Exp Med. 2006; 203: 1235–47.
- Cho JA, Park H, Kim HK, Lim EH, Seo SW, Choi JS et al. Hyperthermia-treated mesenchymal stem cells exert antitumor effects on human carcinoma cell line. Cancer. 2009; 115: 311–23.
- Patel T. Cholangiocarcinoma – controversies and challenges. Nat Rev Gastroenterol Hepatol. 2011; 8: 189–200.
- Sirica AE, Dumur CI, Campbell DJ, Almenara JA, Ogunwobi OO, Dewitt JL. Intrahepatic cholangiocarcinoma progression: prognostic factors and basic mechanisms. Clin Gastroenterol Hepatol. 2009; 7: S68–78.
- Sirica AE. The role of cancer-associated myofibroblasts in intrahepatic cholangiocarcinoma. Nat Rev Gastroenterol Hepatol. 2012; 9: 44–54.
- Kajiyama K, Maeda T, Takenaka K, Sugimachi K, Tsuneyoshi M. The significance of stromal desmoplasia in intrahepatic cholangiocarcinoma: a special reference of ‘scirrhous-type’ and ‘nonscirrhous-type’ growth. Am J Surg Pathol. 1999; 23: 892–902.
- Sirica AE, Campbell DJ, Dumur CI. Cancer-associated fibroblasts in intrahepatic cholangiocarcinoma. Curr Opin Gastroenterol. 2011; 27: 276–84.
- Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell. 2010; 18: 884–901.
- Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009; 9: 581–93.
- Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008; 10: 1470–6.
- Saunderson SC, Schuberth PC, Dunn AC, Miller L, Hock BD, MacKay PA et al. Induction of exosome release in primary B cells stimulated via CD40 and the IL-4 receptor. J Immunol. 2008; 180: 8146–52.
- Kogure T, Lin WL, Yan IK, Braconi C, Patel T. Intercellular nanovesicle-mediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology. 2011; 54: 1237–48.
- Gutwein P, Stoeck A, Riedle S, Gast D, Runz S, Condon TP et al. Cleavage of L1 in exosomes and apoptotic membrane vesicles released from ovarian carcinoma cells. Clin Cancer Res. 2005; 11: 2492–501.
- Kogure T, Yan I, Lin W, Patel T. Extracellular vesicle–mediated transfer of a novel long noncoding RNA TUC339: a mechanism of intercellular signaling in human hepatocellular cancer. Genes Cancer. 2013; 4: 261–72.
- Marienfeld C, Tadlock L, Yamagiwa Y, Patel T. Inhibition of cholangiocarcinoma growth by tannic acid. Hepatology. 2003; 37: 1097–104.
- Nakahara H, Dennis JE, Bruder SP, Haynesworth SE, Lennon DP, Caplan AI. In vitro differentiation of bone and hypertrophic cartilage from periosteal-derived cells. Exp Cell Res. 1991; 195: 492–503.
- Lazarus HM, Koc ON, Devine SM, Curtin P, Maziarz RT, Holland HK et al. Cotransplantation of HLA-identical sibling culture-expanded mesenchymal stem cells and hematopoietic stem cells in hematologic malignancy patients. Biol Blood Marrow Transplant. 2005; 11: 389–98.
- Kogure T, Patel T. Isolation of extracellular nanovesicle microRNA from liver cancer cells in culture. Methods Mol Biol. 2013; 1024: 11–18.
- Yamagiwa Y, Marienfeld C, Tadlock L, Patel T. Translational regulation by p38 mitogen-activated protein kinase signaling during human cholangiocarcinoma growth. Hepatology. 2003; 38: 158–66.
- Wehbe H, Henson R, Meng F, Mize-Berge J, Patel T. Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res. 2006; 66: 10517–24.
- Park J, Tadlock L, Gores GJ, Patel T. Inhibition of interleukin 6-mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology. 1999; 30: 1128–33.
- Braconi C, Patel T. Cholangiocarcinoma: new insights into disease pathogenesis and biology. Infect Dis Clin North Am. 2010; 24: 871–84, vii.
- Prockop DJ, Oh JY. Mesenchymal stem/stromal cells (MSCs): role as guardians of inflammation. Mol Ther. 2012; 20: 14–20.
- Zischek C, Niess H, Ischenko I, Conrad C, Huss R, Jauch KW et al. Targeting tumor stroma using engineered mesenchymal stem cells reduces the growth of pancreatic carcinoma. Ann Surg. 2009; 250: 747–53.
- Hannafon BN, Ding WQ. Intercellular communication by exosome-derived microRNAs in cancer. Int J Mol Sci. 2013; 14: 14240–69.
- Gu J, Qian H, Shen L, Zhang X, Zhu W, Huang L et al. Gastric cancer exosomes trigger differentiation of umbilical cord derived mesenchymal stem cells to carcinoma-associated fibroblasts through TGF-beta/Smad pathway. PLoS One. 2012; 7: e52465.
- Cho JA, Park H, Lim EH, Lee KW. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int J Oncol. 2012; 40: 130–8.
- Cho JA, Park H, Lim EH, Kim KH, Choi JS, Lee JH et al. Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts. Gynecol Oncol. 2011; 123: 379–86.
- Tsai KS, Yang SH, Lei YP, Tsai CC, Chen HW, Hsu CY et al. Mesenchymal stem cells promote formation of colorectal tumors in mice. Gastroenterology. 2011; 141: 1046–56.
- Shinagawa K, Kitadai Y, Tanaka M, Sumida T, Kodama M, Higashi Y et al. Mesenchymal stem cells enhance growth and metastasis of colon cancer. Int J Cancer. 2010; 127: 2323–33.
- De Boeck A, Pauwels P, Hensen K, Rummens JL, Westbroek W, Hendrix A et al. Bone marrow-derived mesenchymal stem cells promote colorectal cancer progression through paracrine neuregulin 1/HER3 signalling. Gut. 2013; 62: 550–60.
- De Boeck A, Narine K, De Neve W, Mareel M, Bracke M, De Wever O. Resident and bone marrow-derived mesenchymal stem cells in head and neck squamous cell carcinoma. Oral Oncol. 2010; 46: 336–42.
- Liu J, Han G, Liu H, Qin C. Suppression of cholangiocarcinoma cell growth by human umbilical cord mesenchymal stem cells: a possible role of Wnt and Akt signaling. PLoS One. 2013; 8: e62844.
- Klopp AH, Gupta A, Spaeth E, Andreeff M, Marini F. Concise review: dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth?. Stem Cells. 2011; 29: 11–19.
- Tadlock L, Patel T. Involvement of p38 mitogen-activated protein kinase signaling in transformed growth of a cholangiocarcinoma cell line. Hepatology. 2001; 33: 43–51.
- Braconi C, Swenson E, Kogure T, Huang N, Patel T. Targeting the IL-6 dependent phenotype can identify novel therapies for cholangiocarcinoma. PLoS One. 2010; 5: e15195.
- Tu B, Du L, Fan QM, Tang Z, Tang TT. STAT3 activation by IL-6 from mesenchymal stem cells promotes the proliferation and metastasis of osteosarcoma. Cancer Lett. 2012; 325: 80–8.
- Huang WH, Chang MC, Tsai KS, Hung MC, Chen HL, Hung SC. Mesenchymal stem cells promote growth and angiogenesis of tumors in mice. Oncogene. 2013; 32: 4343–54.
- Owen JD, Strieter R, Burdick M, Haghnegahdar H, Nanney L, Shattuck-Brandt R et al. Enhanced tumor-forming capacity for immortalized melanocytes expressing melanoma growth stimulatory activity/growth-regulated cytokine beta and gamma proteins. Int J Cancer. 1997; 73: 94–103.
- Haghnegahdar H, Du JG, Wang DZ, Strieter RM, Burdick MD, Nanney LB et al. The tumorigenic and angiogenic effects of MGSA/GRO proteins in melanoma. J Leukocyte Biol. 2000; 67: 53–62.