Abstract
The gut microbiota consists of trillions of prokaryotes that reside in the intestinal mucosa. This long-established commensalism indicates that these microbes are an integral part of the eukaryotic host. Recent research findings have implicated the dynamics of microbial function in setting thresholds for many physiological parameters. Conversely, it has been convincingly argued that dysbiosis, representing microbial imbalance, may be an important underlying factor that contributes to a variety of diseases, inside and outside the gut. This review discusses the latest findings, including enterotype classification, changes brought on by dysbiosis, gut inflammation, and metabolic mediators in an attempt to underscore the importance of the gut microbiota for human health. A cautiously optimistic idea is taking hold, invoking the gut microbiota as a medium to track, target and treat a plethora of diseases.
In 1976, Micheal Andrews published a book entitled ‘The life that lives on Man’ Citation1, in that he describes with graphic illustrations the array of multicellular life that thrives on the human being. Representing ticks, mites, and other ectoparasites, Andrews’ work reminds us that we and these inhabitants have coevolved and for most parts live peacefully together. They are our ‘intimate companions’.
Within this category of close friends is the multitude of bacteria that live in our gastrointestinal (GI) tract. In humans, the GI tract (a.k.a. the gut) becomes rapidly colonized upon birth, as do other parts of the body. Microbial presence in the GI tract has been much studied not least because the mucosal surface of the human gut affords more than 100 m2 of inhabitable space. The gut microbial community is one of the most densely populated, and the abundance and identities of bacterial species living in the GI tract are now being charted after being pursued for decades () Citation2. The Human Microbiome Project has already highlighted the identification of approximately 30% of the known human gut microflora. A close second at roughly 26% is the resident microflora in the oral cavity Citation3. All in all, humans harbor trillions of bacteria living in tolerant symbiosis. These bacteria, collectively known as microbiota, contribute to an array of host physiological processes. The constituents of the microbiota, ranging from bacterial genes to proteins and metabolites, are collectively referred to as the microbiome. We, humans, are effectively superorganisms, in part governed by our resident microbiota. Symbiosis, commensalism, and mutualism prevail in this host–microbe intercourse, and discordance in this marriage is frequently detrimental to the host.
Figure 1. A schematic representation of the lower intestinal tract showing common bacteria found in various parts of the GI tract and bacterial abundance in cfu/ml. Main intestinal functions and pH values found along the GI tract are also shown.(cfu – colony forming unit).
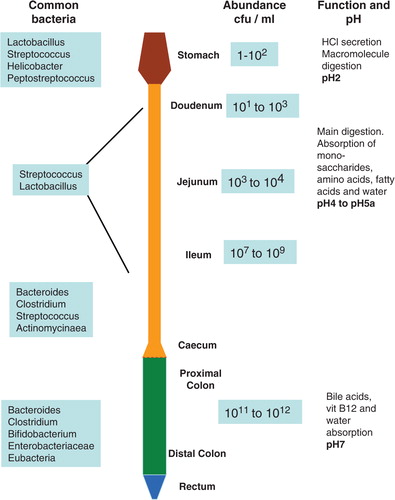
Changes in the gut flora (dysbiosis) and the response of the host to the microbiome have been linked to diseases such as obesity and chronic inflammation. Being naturally well placed, the gut microbiota has been postulated to contribute to energy harvest, a process that occurs in the intestine. We will discuss the complexity surrounding the role of the gut flora in the current obesity epidemic. This review will also aim to explore the latest findings that place the microbiota within reach of the classical host–microbe defense and gut homeostasis. Additional insight into other physiological parameters that may benefit from microbial input will be briefly highlighted.
Microbial composition and functions in the GI tract
In recent years, concerted efforts to identify, describe, and quantify the bacterial communities of the mammalian gastrointestinal tract have begun to bear fruit. Many international consortia and collaborations have been established to answer the question: ‘who is there?’ Hot on the heels of this is the query: ‘what are they doing there?’ This second, crucial, and rather more complex question is yet to be satisfactorily tackled, but fascinating glimpses of what lies ahead have been caught. The Human Microbiome Project in the USA and the Metagenomics of the Human Intestinal Tract (MetaHIT) consortium in Europe are only two of the major initiatives with the purpose of characterizing the microbial communities that inhabit multiple sites in the human body (skin, nasal, oral, urogenital, and intestinal flora) and to look for correlations between the changes in the microbiome and prevalence of diseases. Collaborations to investigate if there is a common core of bacterial species shared among all or the majority of human beings have also been initiated, resulting in some interesting paradigms.
In the uterus, the developing fetus is believed to be completely devoid of bacterial flora. The same cannot be said of bacterial influence on the growing fetus. Upon birth, the intestinal tract of the infant begins to be colonized, ultimately creating a rich and diverse microenvironment. The microbiota is first acquired in the birth canal during delivery and then through breast milk. Fecal microbial profiles of infants show a striking similarity to maternal vaginal and breast milk bacterial profiles Citation4. During the course of early childhood, microbial composition changes with age and diet Citation5 Citation6. From the evolutionary point of view, the character of gut microbiota strongly depends on the main nutritional source, separating bacterial profiles of omnivores, carnivores, and herbivores Citation7
Several groups have been studying the convergence and variability of the human gut microbiome. These studies were normally performed on a small group of healthy individuals. However, even though the sample size has been minimal, there were some major accomplishments, setting the scene for further large-scale metagenomic approaches. One initial observation was that the human gut flora belongs mainly to only two phyla – Firmicutes (mostly represented by Clostridia) and Bacteroidetes, with a smaller representation of bacteria belonging to Proteobacteria and Actinobacteria Citation8. The second observation brought about the discovery of Archaea, mostly represented by the methanogenic Methanobrevibacter smithii Citation8 Citation9. M. smithi facilitates fermentation by reducing the acidicity of the lower intestine. What is interesting is that the diversity of Archaea is but a fraction of the expansiveness of the bacterial species colonizing the gut. Nevertheless, it has been argued that Archaea may yet contribute to the health status of the human gut, due to the unique nature of their metabolic prowess Citation10.
The composition of the bacterial flora seems to strongly depend on the environment and the maternal input during delivery and upon breast feeding. In humans, members of the same family were shown to have bacterial communities that were more similar to each other than to unrelated individuals Citation11. Moreover, this feature was host genotype independent because there was no significant difference detected in the degree of similarity between samples collected from mono- and dizigotic twins. This likely suggests that the environment and maternal flora, rather than genetic factors alone, have greater impact on establishing bacterial communities in a new individual. This observation confirmed the statement that the largest amount of variability between the gut bacterial flora could be explained by intersubject differences Citation8.
Recent second generation, high throughput studies have revealed more details about the human bacterial communities. One major study specifically aimed at assessing whether a common bacterial core shared by all or at least a majority of humans could be determined Citation12. Fecal material collected from a large cohort of individuals in different countries was subjected to metagenomic analysis. The bacterial groups that were present in the samples in vast numbers were Bacteroidetes and Dorea/Eubacterium/Ruminococcus. Only 18 species were present in all individuals examined, while 57 species were seen in more than 90% of samples and around 50% shared 75 species. Although these numbers are higher than routinely observed, they are indicative of the diversity of the human microbiome, while at the same time emphasizing the extent of the interindividual differences.
To obtain a better picture of the role or function of the bacterial communities, a handle on bacterial gene expression was obviously necessary. Failing this, concerted efforts have been made to first identify genes that are encoded by intestinal microbiota and to place their relevance within the context of gut function. More than 3 million open reading frames (ORF) of bacterial genes have been, thus, identified, potentially coding for as many genes. However, the majority of these genes could only be found in very few samples, again confirming the huge variability between individuals. Only around 300,000 genes were found in more than half of the individuals queried, denoting commonly shared microbial genes. Each individual subject carried around 500,000 bacterially encoded genes, among which the majority were rare genes, shared by less than half of the group sampled Citation12. Around 2.5% of the microbially encoded genes could be assigned to the functional group of enzymes involved in carbohydrate metabolism, and again these sets of genes were more similar within individuals of the same family Citation11. Attention has also been drawn to the minimal gut microbiome – genes that are present in most bacteria and code for functions that are necessary for a bacterium to survive in the gut. This set of genes can be divided into housekeeping genes (amino acid synthesis, central carbon metabolism, and protein complexes such as RNA/DNA polymerases) and those involved in gut-specific functions (fermentation and adhesion). However, large portions of the ORF/genes were of unknown functions Citation12. On the other hand, efforts have been made to identify the minimal gut metagenome coding for functions involved in the homeostasis of the whole ecosystem, which are present in most individual host samples. Most of these common bacterial genes are involved in the digestion of complex sugars and their subsequent fermentation. These genes were coded not by the most abundant bacteria but rather by species present in low number, suggesting that even a relatively small bacterial group might be crucial for the establishment of a well functioning gut, as exemplified by Archaea. One must, however, note that the minimal gut microbiome and minimal gut metagenome are not mutually exclusive, and there are many genes that will belong to both groups, being necessary both for the survival of the bacterium as well as for gut function. A higher degree of redundancy at the gene level rather than taxonomic level suggests that the core microbiome should exhibit shared bacterial functions that need to operate to ensure survival and successful symbiosis in the gut Citation11.
In a more recent study, inching closer to marrying microbial profiles and defined functions, Arumugam et al. (2011) have proposed a relatively new concept in the classification of the gut microbiome Citation13. In this study, using stool samples representing different ethnicities, countries, and continents, the authors chanced on a perplexingly simple finding: despite a large and dynamic bacterial community, the combination of the microbes in each individual appeared to be systematically organized into clusters termed enterotypes. Even more surprising was that only three enterotypes were identified based on different clusters of bacterial species. Each had varied but defined functional profiles arising from these clusters. A unique set of properties have been assigned to each enterotype, notably that there is no geographical demarcation, and further, no correlation between presence of a given enterotype with sex, age, or body mass index (BMI) could be shown. The three enterotypes can be identified and viewed on various levels – prevalence of bacteria, methods of harvesting energy, and overrepresentation of certain enzymatic pathways. For instance, enterotype 1 is dominated by Bacteroides that derive energy mostly from fermentation of sugars and is enriched in genes coding for biotin biosynthesis. The second enterotype is driven by Prevotella and can be characterized by enrichment of genes coding for thiamine biosynthesis and harvesting energy from biodegradation of mucin glycoproteins. Enterotype 3 is mostly enriched in Ruminococcus, also known to be able to degrade mucins. This enterotype is, however, enriched in genes coding heme biosynthesis in contrast to the Prevotella enterotype. The phylogenetic structure in this instance appears to be indicative of specific functional properties (summarized in ). The contemporary concept of enterotypes was given a boost recently when another study independently confirmed the existence of two enterotypes in a population of 98 individuals Citation6. The study further found that the enterotype was associated with long-term diet as shown by transient feeding experiments. Hence, although significant changes in the microbiota can occur rapidly on dietary change, no temporary shifts in enterotypes are forthcoming, that is the enterotype does not seem to budge easily.
Figure 2. The human gut flora has recently been classified as belonging to one of three different clusters termed enterotypes. This diagram postulates that properties of the microbiome unique to each enterotype (hatched-hexagon, hatched-triangle and hatched-rhombus) give rise to enterotype-specific effects. Each enterotype likely also harbors common essential functions that may then facilitate bacterial survival in the gut. Both enterotype-specific and common functions can collectively impact on host response.
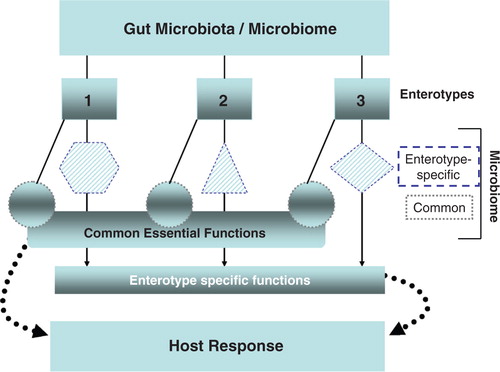
The properties governing the evolution and stabilization of the enterotypes remain to be discovered. The role of the individual's immune system, nutritional input during early childhood, lifestyle, and the exposure to other gut microbial ecologies during the formative years of the microbiome may play a role in the establishment of the individual's enterotype. The use of genetically identical individuals (twin registries), patient cohorts, and materials collected from longitudinal studies may aid in answering some of these queries. Finally, although exciting, the enterotype postulate does require further, robust proof for universal acceptance and use. It is altogether heartening to see that the findings of this study do not preclude the existence of additional enterotypes.
A word of caution
The results obtained from analyzing the bacterial communities of the human gut and their roles in host physiology suggest transience in the gut microbiota, mechanisms of which remain to be discovered Citation4 Citation6. The 16S RNA-based methods of identification still have a number of limitations including incomplete coverage of the sequenced gene fragments and problems of correct alignment to known bacterial genomes. One predictable problem in trying to fuse function and identity is of course aligning the bacterial presence with respective gene expression patterns. This conundrum has yet to be seriously tackled, although limited steps to redress this problem are underway. Gosalbes et al. (2011) in a metatranscriptomic study of 10 volunteers found microbial mRNA expression to have a stable representation of genes involved in nutrient processing, energy harvest, and biosynthesis of cellular components. Lipid and amino acid metabolism were poorly reflected in the expression pattern. Finally, this study also found expression of small RNAs that may have higher regulatory properties. Although in no way conclusive, this study highlights an additional criterion of gut health assessment to complement with the concept of enterotype that is presently available only at the metagenomic level Citation14.
To facilitate rapid, easy, and non-invasive procurement of samples, fecal material is generally used in microbiota profiling. In such studies, the fecal microbiota is used to infer the entire gut microflora, and the lack of information about the potential difference between specific anatomical sites and viability of the bacteria, although glaringly obvious, is usually ignored. One very prudent step in trying to determine ‘active’ microbiota composition has been to compare microbial communities residing on intestinal mucosal surfaces with those found in the feces. The conclusions from such studies point toward variabilities in microbial composition between sample sites, indicating that looking at bacterial communities in the fecal sample may not be as representative for the gut microbiome as we would like to see it Citation8.
The results obtained from fecal-derived studies have, nevertheless, given added credence to the long-held view that different physiological properties may be influenced by different microbial manifestations, for example varied microbiomes or enterotypes. This may lend itself to some plasticity in the bacterial clusters, while retaining core enzymatic and biochemical functions that will reflect the individual enterotypes. The current definition of enterotypes will no doubt undergo further refinement over the next few years. This is likely to provide a better picture of microbial properties that impact on health status, xenobiotic, and nutritional metabolism and perhaps act as an indicator of disease susceptibilities. Ideally, this information may in the future aid the choice of preferred therapeutic modules for selected diseases. Although our current knowledge of enterotypes is far from complete, the potential uses of this new microbiome classification are tantalizing and set a standard to which the disease-associated bacterial flora or microbiome may be compared.
Microbiota and its impact on host metabolism
Nutritional metabolism and xenobiotic metabolism are indispensable cornerstones of survival. The former is a program to extract, preserve, and build energy reserves, whereas the latter will identify and eliminate elements detrimental to the long-term survival of the organism. In superorganisms such as ourselves (and other eukaryotes), the resident microbiota is able to influence both types of metabolic programs. Here, we would like to discuss the current conundrums facing researchers exploring the microbial basis of adipocity. We think that it is wise to discuss the phenomenal progress made thus far, and emphasize the legitimate concerns that still plague the interpretation of the results.
The role of the microbiota in regulation of host energy balance and metabolism can be studied at various levels. By introducing a semi-synthetic high fat diet (HFD), containing a large amount of fat as well as sugars, one can induce weight gain and subsequent development of metabolic diseases Citation15 Citation16 Citation17 . The composition of the bacterial flora can then be monitored simultaneously with changes in the animal's body weight. One very powerful tool to address this question is germ-free mice raised in an environment completely devoid of bacteria. By simply comparing the physiology of germ-free mice with that of conventionally raised animals, one can obtain useful information about how bacteria can shape host metabolism. More interrogative methods rely on using germ-free mice that have been colonized with one specific species of bacteria (monocolonization), a whole group of bacteria, or the entire bacterial flora isolated from conventionally raised animals (coventionalization).
Experiments have shown that germ-free animals seem to be protected from diet-induced obesity Citation18. However, this protective effect was later shown to be strongly dependent on the sugar compositions, that is type of sugar of these diets, not just the amount of fat-derived calories Citation19. This may well be because germ-free animals lack the bacterial enzymes needed to digest polysaccharides, leading, therefore, to a lower calorie intake. Complex polysaccharides are processed in the gut and fermenting microbes produce short-chain fatty acids (SCFAs). Butyrate, propionate, and acetate are SCFAs that can be directly used by colonocytes as an energy source or be further transported to the liver where they can be used as substrates for lipid synthesis Citation20 Citation21. The levels of SCFA can be measured directly in the caecum but more often than not fecal material is used because it is readily available. However, one must remember that neither ceacal nor feacal levels of SCFAs are necessarily true indicators of the amounts produced in the gut because they are continuously used and absorbed by colonocytes. Genes involved in SCFA synthesis were identified in several metagenomic screenings as being overrepresented and highly abundant in the gut flora, strongly suggesting that carbohydrate fermentation is one of the most conserved and important functions of the gut microbiome Citation9 Citation12. Genes governing SCFA biosynthesis were also shown to be stably represented in at least one metatranscriptomic analyses Citation14. Given these observations, a series of experiments were performed to show that the ability of carbohydrate fermentation together with the production of SCFAs was linked to induction of obesity. When germ-free mice were colonized either by whole bacterial flora Citation22 or by saccharolitic fermenting bacteria such as Bacteroidetes thetaiotamicron together with M. smithii, which facilitates fermentation Citation23, an increase in body weight and adiposity was observed. The effect of fermenters on host adiposity was strongly reduced in mice lacking the major SCFA receptor, Gpr41–/– (G-protein coupled receptor 41), hence suggesting a link between SCFA producer and host cell signaling Citation23. In other studies, introduction of HFD in conventionally raised animals was accompanied by a shift toward fermenters in gut flora. This manifested as the appearance of bacterial genes taking part in absorption, transportation, and utilization of simple sugars as well as polysaccharide fermentation leading to SCFA production. The authors then referred to this phenomenon as an ‘obesity-associated microbiome with increased capacity for energy harvest’ Citation24. Animals on HFD had a microbial community that was characterized by a general decrease in microbial diversity and a phylogenetic shift from Bacteroidetes toward Firmicutes Citation24. This could further be attributed to an extensive bloom of one of the families within Firmicutes, namely Mollicutes, belonging to Erysipelotrichaceae Citation25 Citation19. Here, it appears that noticeable phylogenetics shifts need not be a result of changes of many different species but rather the selective overgrowth of one or two members.
The change in the proportional abundance of two major bacterial phyla was then confirmed in another set of experiments. However, in this instance, although the obese mice, as before, had more Firmicutes, the levels of fecal SCFAs were not significantly different Citation26. Intriguingly, one study showing a decrease in fecal SCFA content in obese mice, compared to lean ones, has also been published Citation19. These observations are puzzling and have, thus, been unsatisfactorily addressed. A more detailed approach to determine the pathways and routes of SCFA absorption and use in these mouse models would shed more light on the relationship between HFD, SCFA, and the microbiome.
One of the most intensely studied models of obesity and metabolic syndrome are animals with disrupted leptin signaling. This is achieved in two ways, either by using mice lacking leptin receptor (lep–/– mice) or by deleting the gene for the hormone itself (ob/ob mice). Leptin is crucial for the regulation of food intake (i.e. it induces satiety). Both genotypes of leptin signalling deficiency are characterized by uncontrolled food intake that results in morbidly increased body weight and development of metabolic problems Citation27 Citation28. Comparison of intestinal microbiota compositions of ob/ob mice and their lean wild-type littermates revealed a decrease in Bacteroidetes and a corresponding increase in Firmicutes in obese animals, accompanied by a general decline in microbial diversity in this group Citation29. This shift was probably responsible for the observed increase in abundance of bacterial genes involved in sugar metabolism in another study Citation24. It is tempting to speculate that the increased food intake in those animals favored colonization by bacteria that had a higher metabolic capacity to extract energy, thanks to which both host and microbiota can benefit from otherwise lost calories. Interestingly, the phenotype was somewhat transferable: wild-type germ-free mice colonized with flora from ob/ob mice Citation24 as well as mice that received HFD Citation25 were better at storing fat compared to those colonized with a normal flora.
Apart from effects on host metabolism arising through changes in the gut microbial community or the abundance of metabolites such as short chain fatty acids, the microbiome can also interact with the host tissues by tweaking host gene expression. Among the many recently discovered genes transcriptionally regulated by bacteria is fasting-induced adipose factor (FIAF) also known as angiopoietin-like protein 4 Citation22 Citation30. FIAF is a lipoprotein lipase (LPL) inhibitor that in effect blocks lipid storage mediated through LPL activity. Are et al. 2008 showed that FIAF mRNA in epithelial cells increased in the presence of bacteria such as Enterococcus faecalis Citation30. A putative mechanism through the activity of the metabolically important nuclear receptor peroxisome proliferator-activated receptor gamma (PPARγ) was suggested to govern this increase. In a recent study, increases in the serum level of FIAF caused by administration of Lactobacillus paracasei ssp paracasei F19 were associated with reduced weight gain in HFD-fed mice Citation31. However, other studies reported lowered levels of mRNA expression of FIAF in the intestinal tissue on bacterial exposure Citation22 Citation19. These seemingly confounding results may be explained as reflecting two different situations: Citation1 in which exposure of mice to the whole bacterial community results in a decrease in intestinal FIAF levels; Citation2, while introduction of a single defined bacterial species has the opposite result. Because E. faecalis Citation30 or F19 Citation31 seem to be able to upregulate FIAF expression, it is, therefore, tempting to speculate that certain bacterial species could have a therapeutical role in weight management. Another probiotic strain Lactobacillus gasseri SBT2055 (LG2055) was shown to impact health in small human cohort study. Volunteers with higher BMI and visceral fat when consuming formula milk containing LG2055 lost weight and had reduced abdominal adiposity. This suggests that it has a positive effect on human metabolic health Citation32. The mechanism(s) of action is unknown, but another study in rodents postulated that milk fermented by LG2005 was able to restrict dietary fat absorption in rat intestine Citation33.
In humans, however, the correlation between changes in the gut flora and weight gain are much less clear because all the confounding factors (sex, age, diet, exercise, living environment, antibiotic treatments, disease history, and finally genetic heterogeneity) have to be taken into account. Some groups claim that the shift from Bacteroidetes toward Firmicutes that was observed in mice was also associated with weight gain in humans Citation34. A later study outrightly presents the opposite finding showing a lower ratio of Firmicutes to Bacteroidetes in overweight subjects Citation35. In other reports, the decrease in the abundance of Bacteroidetes could be confirmed, but no difference in Firmicutes was observed, instead a bloom of Actinobacteria carrying the majority of genes overrepresented in obese volunteers was apparent Citation11. In another study, no significant differences were seen in the proportion of Firmicutes versus Bacteroidetes in relation to BMI Citation36. Schwiertz et al. (2010) also suggested that rather than major phylogenetic shifts, discrete, yet metabolically functional changes at the species level may occur in response to dietary changes similar to that observed for Mollicutes in mice (see above) Citation35.
Presently, we do not fully understand the role of microbiota in the regulation of host adiposity. In mice, the shift from Bacteroidetes toward Firmicutes in experiments with obese animals seems well established. The situation in human looks much more complicated, and it is very difficult to reach a consensus on the influence of a specific group of microbiota on changes in body weight. The variability seen in different studies built on microbial composition may be due to a multitude of factors, including diet, volunteer cohort, duration of study, methods of sample preparation, and storage as well as methods of detection. Additionally, concentrations of bacterial metabolites such as SCFA might vary due to increased microbial production, changes in absorption, shifts in microbial cross feeding patterns, or even the rate of transit (reviewed in Citation37, Citation38). Due to the complex nature of the gut environment, it is quite unlikely that a single factor, such as a phylogenetic change in the microbioata, could be responsible for the development of obesity. Rather, changes in eating habits may favor bacterial species that now have to accommodate to a different nutritional environment. Using germ-free mice, Faith et al. (2011) have attempted to formulate a model to predict microbiota changes that are dependent on diet Citation38. Varying the abundance of 10 defined bacterial species that were introduced to the guts of germ-free mice that in turn received different refined diets (protein, sugars, complex carbohydrate, and fat rich), Faith and coworkers were able to infer a relationship between diet and microbiota. It appeared that in general, bacterial presence relied on availability of a single component in the food, indicating that the bacterial community may predominantly depend on the diet and can be changed by manipulating the type of food ingested. Within this limited study at least, the diet appears to govern gut microbial profile. However, as the authors rightly point out, one challenge ahead is to determine how the complex interplay between the human microbiome and the inordinate variability of the human diet affects parameters of gut health.
Presently, it is difficult to show whether the changes in the bacterial flora preceded weight increase, or if the changes that occur in the presence of each bacterial species are strongly associated with the ingested food. Introducing more processed foods and antibiotics has led and will lead in the future to the reduction in the diversity of the microbial flora, simply because there will be a gradual decrease in nutrients available for microbial use (reviewed in Citation39). It seems likely that a balanced bacterial flora is needed for maintaining good health, and perturbing this balance could disrupt controlled energy harvest leading to problems such as obesity.
The microbiota and inflammatory bowel diseases
Inflammatory Bowel Disease (IBD) is an example of a complex disease postulated to arise following a disharmonious relationship between the immune system and the commensal flora. Genetic, microbial as well as dietary and environmental cues together seem to play a role in the development of pathology. There are two major forms of IBD – Ulcerative Colitis (UC) affecting mostly colon and Crohn's Disease (CD) affecting colon as well as the distal part of the small intestine (reviewed in Citation40, Citation41). Previous studies have demonstrated that intestinal dysbiosis, characterized mainly by altered bacterial composition and loss of diversity, is strongly associated with the development of the disease Citation42 Citation43 Citation44 Citation12. There are two major models for experimental colitis, chemically induced colitis, and spontaneous colitis occurring in genetically modified animals. Two methods to chemically induce colitis are extensively used: the administration of either dextran sulphate sodium (DSS) in drinking water or 2, 4, 6-trinitrobenzenesulfonic acid intrarectally. Both chemically induced and spontaneous models have advantages and disadvantages that have been extensively reviewed elsewhere Citation45.
One posit that perturbed balance between the commensals bacteria and the host may play a role in the development of obesity is equally applicable to the pathogenesis of IBD. Studies performed on animals depleted of certain genes involved in immune regulation of gut homeostasis show that specific genetic mutations harbored by the host may cause an uncontrolled growth of otherwise underrepresented bacteria, which leads to development of colitis (reviewed in Citation46). In recent studies, this altered microbiota was transmitted to wild-type animals that then develop the disease, suggesting that the microbiota retained the disease-causing shift that can be transmitted between individuals (especially from mother to pups) Citation47 Citation48
Epithelial cells have the ability to sense microbes by recognizing the microbe-associated molecular patterns (MAMPs). Several families of receptors expressed both on the cell surface as well as in the cytoplasm are involved in recognition of those MAMP molecules, such as Toll-like receptors (TLRs) Citation49, nucleotide-binding oligomerization domain (NOD), Citation50 and NOD-like receptors (NLRs) Citation51 Citation52. In this review, we would like to briefly highlight the involvement of all three of these recognition molecules in the pathogenesis of IBD, and how this may be connected to shifts in microbial communities.
Recognition of microbiota by TLRs is essential for induction of inflammation and immune response. Ligands recognized by TLRs are not only specific for pathogens but are also present on commensal microbiota, and it remains to be understood how the host distinguishes danger signals and homeostatic signals. Most TLRs use an adaptor molecule MyD88 to transmit the signals within the cell Citation49. MyD88-, TLR2-, and TLR4-deficient mice show a more severe phenotype of DSS-induced colitis compared to their wild-type littermates. Similarly, animals with depleted microbiota (following treatment with antibiotics) show increased susceptibility to experimental colitis. On the other hand, administration of TLR4 or TLR2 ligands, in microbiota-depleted animals, was able to ameliorate the disease to a level observed in animals harboring normal flora. These results strongly suggest that commensal microbiota play an important role in stimulating TLRs and setting the threshold of immune response needed for homeostasis Citation53. Additionally, spontaneous colitis observed in conventionally raised, but not germ-free IL10 –/– mice is dependent on MyD88 signaling. Deletion of MyD88 in IL10 –/– animals abolishes TLR signaling and is protective against the development of the disease. These results suggest that TLR signaling, although necessary for establishing and maintaining balance between pro- and antiinflammatory signals, might also be responsible for development of the disease if other factors, such as IL10, are missing or when the balance is otherwise disrupted Citation54.
In addition to TLRs, microbial molecules can be recognized by members of the NOD family that reside in the cytoplasm – NOD1 and NOD2 Citation50. Mutations in the NOD2 gene have been associated with increased susceptibility to IBD, especially CD, but the precise role of this protein in the pathogenesis of the disease remains unknown Citation55. NOD2 –/– mice display abnormal Paneth cell functions and develop symptoms similar to CD Citation56 Citation57. Microbial samples coming from patients harboring mutations in the NOD2 gene display a shift in bacterial composition not seen in individuals not carrying these mutations Citation58. Additionally, NOD2–/– animals harbor a microflora that is different from that of wild-type animals Citation59.
The third important group of defense molecules is NLRs that form cytoplasmic complexes known as inflammasomes that are able to sense both endo- and exogenic stress or damage-associated signals (reviewed in Citation60). When activated, they coassemble with the adaptor protein – apoptosis-associated speck-like protein (ASC), into multiprotein complexes that activate caspase 1 and subsequent cleavage of IL-1b and IL-18 Citation61 Citation62. Mice that lack components of the inflammasome (NLRP6 –/–, ASC –/–) or downstream targets of active complex (caspase 1 –/–, IL-18–/–) suffer from more severe DSS-induced colitis than their wild-type littermates Citation48. Inflammasome loss resulted in the development of more colitogenic microflora that could be transferred to wild-type mice that were cohoused or cofostered with knock-out mice, resulting in more severe phenotype in these animals. Two groups of bacteria were associated with the colitogenic microbiome: family Prevotellaceae in the phylum Bacteroidetes as well as a genus belonging to TM7 phylum. Both groups are capable of disrupting the mucus barrier through its degradation and have been previously associated with development of IBD and periodontal disease. The Prevotella cluster has been detected on mucosal surfaces taken from patients suffering from UC or CD but not in the guts of healthy subjects Citation63. Lucke et al. 2006 also highlighted the abundance of the members of the Prevotella genera in mucosal biopsy specimens from UC patients Citation64. On the species level, Prevotella denticola is much more prevalent in subjects suffering from periodontitis than in healthy subjects Citation65. Antibiotic resistant bacteria belonging to Prevotella genus has also been identified among periodontal bacteria Citation66. TM7 phylum members (gram-positive, non-cultivable bacteria) could be found in equal numbers in biopsies obtained from UC, CD, and non-IBD patients; however, the composition of the cloned 16S rRNA sequences differed significantly between diseased and healthy samples Citation67. CD patients had a more diverse (broader species spectrum) TM7 flora. One of the clones showed very high similarity to a clone suggested to play a causative role in periodontal disease Citation68, and many members of that phylum have been associated with oral inflammation Citation69 Citation65 Citation70. Hence, there appear to be commonalities in mucosal inflammation, be it manifesting in the oral or intestinal cavities.
Garrett et al. 2010 have identified two species belonging to Enterobacteriaea – Klebsiella pneumoniae and Proteus mirabilis as highly associated with colitogenic phenotype of T-bet–/– RAG2–/– mice Citation71. These animals harbor a deficiency in innate immunity relying on T-cells responses, and they spontaneously develop colitis (referred by authors as T-bet–/– RAG2–/– UC – TRUC mice) Citation47. The colitic phenotype was transmittable to cohoused or cross-fostered WT mice, akin to the inflammasome-deficient mice discussed above Citation71. Neither of the identified species was, however, able to cause colitis when transferred to germ-free mice, indicating a need for other bacterial signals for a full-blown inflammatory reaction. Klebsiella pneumoniae together with other drug-resistant Enterobacteriacae has been previously reported to be present in oral cavities of patients with periodontal disease. It is postulated that the hydrolytic enzymes produced by these bacteria play a role in the destruction of the tooth-supporting tissues Citation72.
Frank et al. (2007) identified additional shifts in microbiota in UC and CD patients compared to non-IBD samples Citation73. UC and CD patients had fewer sequences representative of Bacteroides and Lachnospiraceae (a subgroup of Firmicutes) and an overrepresentation of Proteobacteria and also Bacillus – another subgroup of Firmicutes. In the small intestine, proteobacterial sequences were more abundant and Bacillus sequences less so, in UC and CD patients, relative to non-IBD samples. The bacterial profiles belonging to some CD and UC patients were clustered separately from the non-IBD samples and were characterized among the others by a decreased bacterial load. A reduced bacterial diversity was also observed by Qin et al. (2010) Citation12. IBD patients recruited to this study harbored on average 25% less bacterial genes than healthy subjects, and principal component analysis clearly separated IBD patients from healthy subjects and CD patients from UC patients. One speculation, built on the Hygiene Hypothesis, is that decreasing diversity of the bacterial ecology may be a causative factor in diseases such as IBD.
A new player in IBD pathogenesis is ATG16L1 (Autophagy-related 16-like 1) that codes for a protein that is a part of a large complex necessary for autophagy – a process in which cell components are targeted to degradation. The disease variant of the gene can be found in around 50% of European population and confers a twofold increase in disease susceptibility Citation74 Citation75 Citation76 . People harboring such mutations also have altered microbial flora Citation58. In mice with deleted ATG16L1, a reduced capacity for autophagy is seen Citation77 Citation78. On viral infection with a murine norovirus (MNV), AGT16L1–/– mice display additional features with strikingly abnormal Paneth cells, resulting in aberrant packing and exocytosis of antimicrobial peptides. On DSS treatment, these mice developed colonic ulcers with various hallmarks of CD-associated lesions. Administration of antibiotics in these mice prevented the abnormal DSS-induced pathologies suggesting the necessity of the microflora to induce a phenotype resembling CD Citation79. Interestingly, although antibiotics have been used to treat CD and UC, it seems to be more effective in the case of the former, as they could also prevent the relapse of the disease Citation80.
Apart from the inflammatory disorder, IBD also predisposes to colorectal cancer (CRC). Around 18% of patients will develop CRC after 30 years of disease duration Citation81 Citation82. Changes in bacterial composition seen prior to the manifestation of colitis may also be associated with the pathology of colorectal cancer because TRUC mice (described above) spontaneously develop colonic dysplasia and rectal adenocarcinoma, resembling human colonic cancers associated with IBD Citation83.
So far only one bacterial species – enterotoxigenic Bacillus fragilis could be directly linked to development of CRC. Toxin produced by this bacterium (BFT) was identified as being a causative factor for both colitis and very early tumorigenesis in the APCMin mouse model of colon cancer (harboring a heterozygous mutation in the APC gene) Citation84. BFT acts as an oncogene by cleaving E-cadherin, a tumor suppressor protein triggering the nuclear localization of B-catenin and colonic cells proliferation, through the activation of the Wnt pathway. It also activates the NF-κB signaling pathway in the cells that results in secretion of proinflammatory cytokines. Interestingly, BTF exerts its oncogenic functions via the Th17 cells Citation84, the development of which is solely dependent on the presence of commensal bacteria in the gut Citation85. Hence, the pathologies linked to Klebsiella Citation71 and B. fragilis appear to be dependent on the presence of the whole microbial community.
Although many bacterial species or groups are associated with the pathology of IBD, other prokaryotic partners may play protective roles. Interestingly, B. fragilis implicated earlier in CRC may have protective effects in infectious colitis caused by Helicobacter hepaticus. Interestingly, administration of a single component of B. fragilis cell wall – namely polysaccharide A (PSA) – Citation86 is enough to repress inflammation. Further research could link the protective effects of PSA with the function of IL10 in response to commensal bacteria. PSA was shown to not only prevent but also cure experimental colitis in mice, by actively inducing mucosal tolerance Citation87.
The presence of whole bacterial flora, as well as the fraction of fermenting microbiota, has also been suggested to play a protective role against colitis and helps exclude lethal invasions of pathogenic bacteria. Germ-free animals are characterized by a more severe phenotype of DSS-induced colitis compared to conventionally raised animals Citation88 Citation89. This phenotype could be reverted on conventionalization of GF animals, or by the administration of acetate in drinking water prior to DSS treatment. G-coupled receptor 43 (Gpr43) is one of the cell surface receptors that is activated on binding of acetate. Gpr43–/– mice showed greater morbidity and reduced potential to overcome the disease Citation89. Acetate produced by certain species of Bifidobacteria has also been found recently to protect mice from colitis and subsequent death caused by infection with Escherichia coli O157:H7 Citation90. This clearly indicates that intestinal bacterial metabolites, garnered through microbial processing of available nutrients, do have a significant effect on gut health.
In conclusion, recent studies have prompted researchers to peer deeply into the altered microbial composition in patients suffering from IBD. This may be associated with a general reduction of bacterial diversity as well as shifts toward certain bacterial groups and the appearance of specific, hitherto unidentified species. The changes might be driven by environmental cues and genetic predispositions. As elegantly shown by Cadwell et al. (2010), IBD is a very complex disease and to understand it, we have to look at more than one factor and be able to link microbial changes, presence of viruses with environmental conditions, such as diet and small changes in the host DNA, that are not able to cause a disease when present alone Citation79. This observation only serves to emphasize the importance of understanding the cross talk between different microbial communities thriving in the intestinal milieu. The dynamic communities of intestinal bacterial biofilms are likely to play a major role in setting the stage for host–microbe interactions and subsequent physiological outcomes.
Beyond the intestinal millieu
The vast numbers of bacteria in the gut have over the years been postulated to directly extend their realm of influence beyond the immediate gut environment. Here, we briefly list the role of the gut flora liver function, glucose metabolism, and the control of behavior.
The gut microbiota has been attributed a role in the development of non-alcoholic fatty liver disease (NALFLD) Citation91 Citation92. Urinary metabolites of the NAFLD-susceptible mice strain 129S6 when fed HFD were enriched in microbiota-derived methylamines: dimethylamine, trimethylamine (TMA), and trimethylamine-N-oxide (TMAO). TMA is synthesized exclusively by symbiotic bacteria Citation93 and can be further transformed into TMAO by the microbiota themselves or at least in humans by the liver enzyme FMO3. The production of methylamines by microbiota results in decreased bioavailability of choline for the host and seems to trigger NAFLD in mice. This idea was further explored by Wang et al. (2011), who could link high levels of TMAO in plasma with increased risk for cardiovascular disease in humans Citation92.
The interdependence between microbiota, metabolism, and inflammation as well as the host genotype has also been elegantly shown. Mice lacking TLR5 suffer from spontaneous colitis Citation94 and at the same time have higher body weights Citation95 than their wild-type littermates. Interestingly, the increase in weight could be attributed to higher chow consumption and indeed, the trait disappeared when mice were subjected to antibiotic treatment. The obese phenotype could be transferred from TLR5–/– animals to wild-type germ-free mice through the gut flora, suggesting that microbiota may be a factor responsible for hyperphagia and increased body weight in TLR5–/– mice.
Hyperphagia is also very much associated with satiety signals emanating from the gut and directed to the brain. Increasingly, the gut flora is being recognized as a factor in the regulation of the central nervous system (CNS). Colonization by the gut microbiota may influence brain development and animal behavior. Germ-free mice display increased motor activity and reduced anxiety, compared with conventionally raised ones. This phenotype could be reversed if germ-free animals were exposed to bacteria early in life Citation96. Lactobacillus rhamnosus has been recently shown to possess antiinflammatory properties and has been suggested as a candidate to interfere with the CNS signaling through the vagus nerve, which allows communication between the viscera and the brain. This probiotic was shown to reduce plasma levels of stress-induced corticosterone as well as anxiety- and depression-related behavior. These results strongly suggest that modulation of intestinal microbiota in healthy animals may lead to neurochemical changes affecting animal behavior Citation97.
These examples are telling in that they have attracted the attention of researchers who have traditionally been working away from the gut environment to seriously consider the gut microbial milieu when studying a variety of diseases (see ). The scope of this review unfortunately precludes the discussion of microbiome as a source or target for therapies and treatments of various ailments (reviewed in Citation98)
Figure 3. The gut microbiota can affect a variety of host responses. In this schematic diagram, host genotype, the environment, and diet can all impact on the microbiota and the microbiome. The microbiota exerts its effects on the intestine through bacterial signaling molecules and metabolic products (microbiome). Components of the microbiome can also enter the circulation, be transported to various organs, and potentiate multiple effects in these organs (gray arrow). At the same time, the gut–brain axis can circumvent intestinal absorption and may allow the microbiome to directly affect the brain.
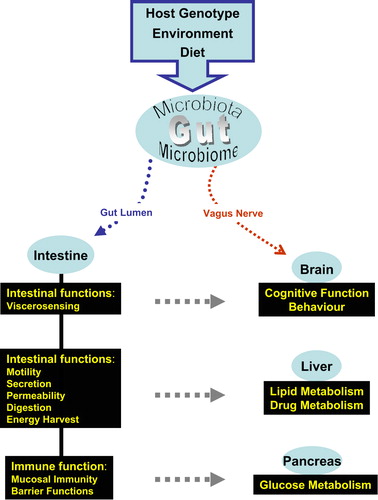
Conclusions
Once touted as the forgotten organ Citation99, the microbiota is slowly beginning to reveal many of its secrets. Having been under close scrutiny for many decades, the gut microbiota is now said to dictate important traits in the human host. Future research delineating properties of the oral microflora should desist from chasing purely metagenomic queries. Bacterial transcriptomics and metabolomics of the oral cavity should instead be earnestly pursued. The oral cavity has one distinct advantage over the GI tract: accessibility, a trait that should be exploited to the fullest. Crucially, information thus accrued would be important to those of us aspiring to understand the workings of the microbiota in the GI tract and beyond.
Painstakingly, slow research of yesterday that lay much of the groundwork for today's ideas has given way to meta-analyses of high throughput findings. Nevertheless, as we begin to answer the questions of ‘who is there’ and ‘what are they doing there,’ we inadvertently arrive at the inquiry ‘how and why’ the superorganism functions. Herein lies the potential to alter the course of many a pathology. What is obvious is the notion that microbial influence permeates into nearly every aspect of our existence. As a component species of a superorganism, we should now universally acknowledge the importance of the microbial world in which we live and, crucially, that which lives on us.
Conflict of interest and funding
There is no conflict of interest in the present study for any of the authors.
Acknowledgements
A.K. was supported by the Cross-Talk FP7-PEOPLE-2007-1-1-ITN-215553 Program. V.A. was funded through the EU project TORNADO (Framework 7, grant no. 222720–2) and by DiabetesFonden, Sweden.
References
- Andrews M. Life that lives on man. London: Faber & Faber. 1976.
- Savage DC. Microbial ecology of the gastrointestinal tract. Ann Rev Microb. 1977; 31: 107–133.
- Peterson J, Garges S, Giovanni M, et al.. The NIH uuman microbiome project. Genome Res. 2009; 19: 2317–323.
- Palmer C, Bik EM, DiGiulio DB, et al.. Development of the human infant intestinal microbiota. PLoS Biol. 2007; 5: e177.
- De Filippo C, Cavalieri D, Di Paola M, et al.. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Nat Acad Sci USA. 2010; 107: 14691–6.
- Wu GD, Chen J, Hoffmann C, et al.. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011; 334: 105–8.
- Ley RE, Hamady M, Lozupone C, et al.. Evolution of mammals and their gut microbes. Science. 2008; 320: 1647–1.
- Eckburg PB, Bik EM, Bernstein CN, et al.. Diversity of the human intestinal microbial flora. Science. 2005; 308: 1635–8.
- Gill SR, Pop M, Deboy RT, et al.. Metagenomic analysis of the human distal gut microbiome. Science. 2006; 312: 1355–9.
- Horz HP, Conrads G. The discussion goes on: what is the role of Euryarchaeota in humans?. Archea. 2010; 2010: 967271.
- Turnbaugh PJ, Hamady M, Yatsunenko T, et al.. A core gut microbiome in obese and lean twins. Nature. 2009; 457: 480–4.
- Qin J, Li R, Raes J, et al.. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010; 464: 59–65.
- Arumugam M, Raes J, Pelletier E, et al.. Enterotypes of the human gut microbiome. Nature. 473: 174–80.
- Gosalbes MJ, Durban A, Pignatelli M, et al.. Metatranscriptomic approach to analyze the functional human gut microbiota. PloS One 6. : e17447.
- Surwit RS, Kuhn CM, Cochrane C, et al.. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988; 37: 1163–67.
- West DB, Boozer CN, Moody DL, et al.. Dietary obesity in nine inbred mouse strains. Amer J Phys. 1992; 262: R1025–32.
- Klaus S. Increasing the protein: carbohydrate ratio in a high-fat diet delays the development of adiposity and improves glucose homeostasis in mice. J Nutr. 2005; 135: 1854–8.
- Backhed F, Manchester JK, Semenkovich CF, et al.. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Nat Acad Sci USA. 2007; 104: 979–84.
- Fleissner CK, Huebel N, Abd El-Bary MM, et al.. Absence of intestinal microbiota does not protect mice from diet-induced obesity. Brit J Nutr. 104: 919–29.
- Wolever TM, Spadafora P, Eshuis H. Interaction between colonic acetate and propionate in humans. Amer J Clin Nutr. 1991; 53: 681–7.
- Scheppach W. Effects of short chain fatty acids on gut morphology and function. Gut. 1994; 35: S35–38.
- Backhed F, Ding H, Wang T, et al.. The gut microbiota as an environmental factor that regulates fat storage. Proc Nat Acad Sci USA. 2004; 101: 15718–3.
- Samuel BS, Shaito A, Motoike T, et al.. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Nat Acad Sci USA. 2008; 105: 16767–2.
- Turnbaugh PJ, Ley RE, Mahowald MA, et al.. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006; 444: 1027–31.
- Turnbaugh PJ, Backhed F, Fulton L, et al.. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008; 3: 213–23.
- Murphy EF, Cotter PD, Healy S, et al.. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut. 2010; 59: 1635–42.
- Tartaglia LA, Dembski M, Weng X, et al.. Identification and expression cloning of a leptin receptor. OB-R. Cell. 1995; 83: 1263–71.
- Zhang Y, Proenca R, Maffei M, et al.. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994; 372: 425–32.
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Nat Acad Sci USA. 2005; 102: 11070–5.
- Are A, Aronsson L, Wang S, et al.. Enterococcus faecalis from newborn babies regulate endogenous PPARgamma activity and IL-10 levels in colonic epithelial cells. Proc Nat Acad Sci USA. 2008; 105: 1943–8.
- Aronsson L, Huang Y, Parini P, et al.. Decreased fat storage by Lactobacillus paracasei is associated with increased levels of angiopoietin-like 4 protein (ANGPTL4). PloS One. 2010; 5: e13087.
- Kadooka Y, Sato M, Imaizumi K, et al.. Regulation of abdominal adiposity by probiotics (Lactobacillus gasseri SBT2055) in adults with obese tendencies in a randomized controlled trial. Eur J Clin Nutr. 2010; 64: 636–43.
- Hamad EM, Sato M, Uzu K, et al.. Milk fermented by Lactobacillus gasseri SBT2055 influences adipocyte size via inhibition of dietary fat absorption in Zucker rats. Brit J Nutr. 2009; 101: 716–24.
- Ley RE, Turnbaugh PJ, Klein S, et al.. Microbial ecology: human gut microbes associated with obesity. Nature. 2006; 444: 1022–23.
- Schwiertz A, Taras D, Schafer K, et al.. Microbiota and SCFA in lean and overweight healthy subjects. Obesity. 2010; 18: 190–95.
- Duncan SH, Lobley GE, Holtrop G, et al.. Human colonic microbiota associated with diet, obesity and weight loss. International J Obesity ; 32. 2005; 2008: 1720–24.
- Velazquez OC, Lederer HM, Rombeau JL. Butyrate and the colonocyte. Production, absorption, metabolism, and therapeutic implications. Adv Exp Med Biol. 1997; 427: 123–34.
- Flint HJ, Bayer EA, Rincon MT, et al.. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nature Reviews. 2008; 6: 121–31.
- Faith JJ, McNulty NP, Rey FE, et al.. Predicting a human gut microbiota's response to diet in gnotobiotic mice. Science. 2011; 333: 101–4.
- Podolsky DK. Inflammatory bowel disease. New Engl J Med. 2002; 347: 417–29.
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007; 448: 427–34.
- Ott SJ, Musfeldt M, Wenderoth DF, et al.. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004; 53: 685–93.
- Manichanh C, Rigottier-Gois L, Bonnaud E, et al.. Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut. 2006; 55: 205–11.
- Seksik P, Sokol H, Lepage P, et al.. Review article: the role of bacteria in onset and perpetuation of inflammatory bowel disease. Aliment Pharm Therap. 2006; 24: 11–18.
- Hoffmann JC, Pawlowski NN, Kuhl AA, et al.. Animal models of inflammatory bowel disease: an overview. Pathobiology. 2002; 70: 121–30.
- Saleh M, Elson CO. Experimental inflammatory bowel disease: insights into the host-microbiota dialog. Immunity. 2011; 34: 293–302.
- Garrett WS, Lord GM, Punit S, et al.. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007; 131: 33–45.
- Elinav E, Strowig T, Kau AL, et al.. 2 NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011; 145: 745–57.
- Takeda K, Kaisho T, Akira S. Toll-like receptors. Ann Rev Immunol. 2003; 21: 335–76.
- Inohara N. Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nature Revs. 2003; 3: 371–82.
- Franchi L, McDonald C, Kanneganti TD, et al.. Nucleotide-binding oligomerization domain-like receptors: intracellular pattern recognition molecules for pathogen detection and host defense. J Immunol. 2006; 177: 3507–513.
- Kaparakis M, Philpott DJ, Ferrero RL. Mammalian NLR proteins; discriminating foe from friend. Immunol. Cell Biol. 2007; 85: 495–502.
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, et al.. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004; 118: 229–41.
- Rakoff-Nahoum S, Hao L, Medzhitov R. Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity. 2006; 25: 319–29.
- Hugot JP, Chamaillard M, Zouali H, et al.. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001; 411: 599–603.
- Ogura Y, Lala S, Xin W, et al.. Expression of NOD2 in Paneth cells: a possible link to Crohn's ileitis. Gut. 2003; 52: 1591–7.
- Wehkamp J, Harder J, Weichenthal M, et al.. NOD2 (CARD15) mutations in Crohn's disease are associated with diminished mucosal alpha-defensin expression. Gut. 2004; 53: 1658–64.
- Frank DN, Robertson CE, Hamm CM, et al.. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2011; 17: 179–84.
- Rehman A, Sina C, Gavrilova O, et al.. Nod2 is essential for temporal development of intestinal microbial communities. Gut. 2011; 60: 1354–62.
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010; 140: 821–32.
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002; 10: 417–26.
- Agostini L, Martinon F, Burns K, et al.. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004; 20: 319–25.
- Kleessen B, Kroesen AJ, Buhr HJ, et al.. Mucosal and invading bacteria in patients with inflammatory bowel disease compared with controls. Scan J Gastroentero. 2002; 37: 1034–41.
- Lucke K, Miehlke S, Jacobs E, et al.. Prevalence of Bacteroides and Prevotella spp. in ulcerative colitis. J Med Microb. 2006; 55: 617–24.
- Kumar PS, Griffen AL, Barton JA, et al.. New bacterial species associated with chronic periodontitis. J Dent Res. 2003; 82: 338–44.
- Van Winkelhoff AJ, Winkel EG, Barendregt D, et al.. Beta-lactamase producing bacteria in adult periodontitis. J Clin Periodontol. 1997; 24: 538–43.
- Kuehbacher T, Rehman A, Lepage P, et al.. Intestinal TM7 bacterial phylogenies in active inflammatory bowel disease. J Med Microb. 2008; 57: 1569–76.
- Brinig MM, Lepp PW, Ouverney CC, et al.. Prevalence of bacteria of division TM7 in human subgingival plaque and their association with disease. Appl Environ Microb. 2003; 69: 1687–94.
- Ouverney CC, Armitage GC, Relman DA. Single-cell enumeration of an uncultivated TM7 subgroup in the human subgingival crevice. Appl Environ Microb. 2003; 69: 6294–98.
- Marcy Y, Ouverney C, Bik EM, et al.. Dissecting biological dark matter with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc Nat Acad Sci USA. 2007; 104: 11889–94.
- Garrett WS, Gallini CA, Yatsunenko T, et al.. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010; 8: 292–300.
- Goncalves MO, Coutinho-Filho WP, Pimenta FP, et al.. Periodontal disease as reservoir for multi-resistant and hydrolytic enterobacterial species. Lett Appl Microbiol. 2007; 44: 488–94.
- Frank DN, St Amand AL, Feldman RA, et al.. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Nat Acad Sci USA. 2007; 104: 13780–5.
- The Wellcome Trust Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007; 447: 661–78.
- Rioux JD, Xavier RJ, Taylor KD, et al.. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nature Genetics. 2007; 39: 596–604.
- Hampe J, Franke A, Rosenstiel P, et al.. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nature Genetics. 2007; 39: 207–11.
- Cadwell K, Liu JY, Brown SL, et al.. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008; 456: 259–63.
- Ju JS, Miller SE, Jackson E, et al.. Quantitation of selective autophagic protein aggregate degradation in vitro and in vivo using luciferase reporters. Autophagy. 2009; 5: 511–9.
- Cadwell K, Patel KK, Maloney NS, et al.. Virus-plus-susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell. 2010; 141: 1135–45.
- Khan KJ, Ullman TA, Ford AC, et al.. Antibiotic therapy in inflammatory bowel disease: a systematic review and meta-analysis. Amer J Gastroenterol. 2011; 106: 661–73.
- Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001; 48: 526–35.
- Xie J, Itzkowitz SH. Cancer in inflammatory bowel disease. World J Gastroentero. 2008; 14: 378–89.
- Garrett WS, Punit S, Gallini CA, et al.. Colitis-associated colorectal cancer driven by T-bet deficiency in dendritic cells. Cancer Cell. 2009; 16: 208–19.
- Wu S, Rhee KJ, Albesiano E, et al.. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nature medicine. 2009; 15: 1016–22.
- Ivanov II, Frutos Rde L, Manel N, et al.. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008; 4: 337–49.
- Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008; 453: 620–5.
- Round JL\, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Nat Acad Sci USA. 2010; 107: 12204–9.
- Kitajima S, Morimoto M, Sagara E, et al.. Dextran sodium sulfate-induced colitis in germ-free IQI/Jic mice. Exp Anim. 2001; 50: 387–95.
- Maslowski KM, Vieira AT, Ng A, et al.. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009; 461: 1282–6.
- Fukuda S, Toh H, Hase K, et al.. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 469: 543–7.
- Dumas ME, Barton RH, Toye A, et al.. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Nat Acad Sci USA. 2006; 103: 12511–6.
- Wang Z, Klipfell E, Bennett BJ, et al.. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011; 472: 57–63.
- Al-Waiz M, Mikov M, Mitchell SC, et al.. The exogenous origin of trimethylamine in the mouse. Metabolism. 1992; 41: 135–6.
- Vijay-Kumar M, Sanders CJ, Taylor RT, et al.. Deletion of TLR5 results in spontaneous colitis in mice. The J.Clin. Invest. 2007; 117: 3909–21.
- Vijay-Kumar M, Aitken JD, Carvalho FA, et al.. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010; 328: 228–31.
- Heijtz RD, Wang S, Anuar F, et al.. Normal gut microbiota modulates brain development and behavior. Proc Nat Acad Sci USA. 108: 3047–52.
- Bravo JA, Forsythe P, Chew MV, et al.. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Nat Acad Sci USA. 2011; 108: 16050–5.
- Cani PD, Delzenne NM. The gut microbiome as therapeutic target. Pharmacol Therapeut. 130: 202–12.
- O'Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Reports. 2006; 7: 688–93.