
Abstract
Background
Reports on the composition of oral bacteriome in Arabs are lacking. In addition, the majority of previous studies on other ethnic groups have been limited by low-resolution taxonomic assignment of next-generation sequencing reads. Furthermore, there has been a conflict about the existence of a ‘core’ bacteriome.
Objective
The objective of this study was to characterize the healthy core oral bacteriome in a young Arab population at the species level.
Methods
Oral rinse DNA samples obtained from 12 stringently selected healthy young subjects of Arab origin were pyrosequenced (454's FLX chemistry) for the bacterial 16S V1–V3 hypervariable region at an average depth of 11,500 reads. High-quality, non-chimeric reads ≥380 bp were classified to the species level using the recently described, prioritized, multistage assignment algorithm. A core bacteriome was defined as taxa present in at least 11 samples. The Chao2, abundance-based coverage estimator (ACE), and Shannon indices were computed to assess species richness and diversity.
Results
Overall, 557 species-level taxa (211±42 per subject) were identified, representing 122 genera and 13 phyla. The core bacteriome comprised 55 species-level taxa belonging to 30 genera and 7 phyla, namely Firmicutes, Proteobacteria, Actinobacteria, Bacteroidetes, Fusobacteria, Saccharibacteria, and SR1. The core species constituted between 67 and 87% of the individual bacteriomes. However, the abundances differed by up to three orders of magnitude among the study subjects. On average, Streptococcus mitis, Rothia mucilaginosa, Haemophilus parainfluenzae, Neisseria flavescence/subflava group, Prevotella melaninogenica, and Veillonella parvula group were the most abundant. Streptococcus sp. C300, a taxon never reported in the oral cavity, was identified as a core species. Species richness was estimated at 586 (Chao2) and 614 (ACE) species, whereas diversity (Shannon index) averaged at 3.99.
Conclusions
A species-level core oral bacteriome representing the majority of reads was identified, which can serve as a reference for comparison with oral bacteriomes of other populations as well as those associated with disease.
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
All microorganisms in the oral cavity and their collective genomes are referred to as oral microbiome (Citation1) – more accurately ‘oral bacteriome’ when only the bacterial component is being described (Citation2). The oral cavity is a home for as many as 1,000 bacterial species (Citation3). They form complex yet stable microbial communities that colonize tissue surfaces in mutualistic relationship with the host. Under certain circumstances, however, the microbial community homeostasis may be lost resulting in overgrowth of a minority of pathogenic species implicated as etiological agents of several oral infectious diseases, the most common being dental caries and periodontitis (Citation4). A proper understanding of the oral bacteriome composition in health is, therefore, the first step toward elucidating the microbial shifts associated with disease. Fortunately, comprehensive characterization of the human microbiome has become feasible with the advent of next-generation sequencing (NGS) technologies that enable analysis of microbiological samples at unprecedented depth and breadth (Citation5).
Several studies have recently used NGS to profile health-associated microbiota in different habitats of the oral cavity including saliva, mucosa, and dental biofilm (Citation6–Citation17). These studies differed in terms of sampling, sequencing technology, the 16S rRNA hypervariable region targeted, and data processing, which partially explains the variations in the results among them. However, there are indeed significant genuine intra-individual, inter-individual, and inter-ethnicity differences in the composition of healthy oral bacteriome (Citation10–Citation14, Citation18). This has led to the evolvement of the concept of core oral bacteriome, which refers to bacterial species shared by the majority of individuals (e.g. 90–100%) (Citation10, Citation13). For example, it is now well recognized that the vast majority of oral bacteriome in all individuals is confined to the five phyla – Firmicutes, Proteobacteria, Actinobacteria, Bacteroidetes, and Fusobacteria – and to a few genera including Streptococcus, Rothia, Neisseria, Prevotella, Haemophilus, Fusobacterium, Porphyromonas, Veillonella, Granulicatella, and Gemella (Citation7, Citation9–Citation11, Citation15) .
One common limitation to the majority of previous studies, however, has been the low taxonomic resolution; that is, the inability to characterize the core oral bacteriome at the strict species level. A typical bioinformatic analysis approach involves de novo clustering of reads into operational taxonomic units (OTUs) followed by using a Bayesian classifier to obtain consensus OTU taxonomic labels. This approach often results in classification of the majority of reads to the genus level (Citation19), which may not be sufficient for characterizing distinct core bacteriomes that are unique to certain populations or diseases. Although including the Human Oral Microbiome Database (HOMD; www.homd.org) (Citation20) as a well-curated, habitat-specific reference set in the analysis improves the resolution of consensus taxonomy assignment, as shown in a few recent studies (Citation16, Citation21) (Citation22), a significant proportion of OTUs remain unclassified at the species level.
In a recent study, we described a robust, species-level read assignment algorithm that searches individual reads for highest sequence similarities to 16S rRNA gene sequences in modified versions of three reference data sets: HOMD, HOMD-extended, and Greengene Gold (GGG) (Citation23). In the current study, we use this algorithm to characterize, at the species level, the core oral bacteriome in a healthy young Arab population.
Methods
Recruitment of study subjects
Twelve subjects (eight males and four females) fulfilling the following criteria were recruited from among students, staff, and patients attending the College of Dentistry, Jazan University: aged 25–35 years; reported Arab origin with two ancestor generations living in the region; no history of tobacco or qat (Catha edulis) use; no history of antibiotic, antifungal, or steroids contraceptives intake in the 3 months prior to sampling; no recent periodontal treatment including prophylaxis; no history of diabetes, immunodeficiency, or pregnancy; no clinical signs of moderate/severe gingivitis or periodontitis; and no visible cavities or extensive fillings. The study was approved by the Ethical Committee for Biomedical Research at Jazan University; all study subjects gave written consent to participate in the study. The characteristics of study population are shown in .
Table 1. Characteristics of the study subjects
Collection and preparation of samples
Mouth rinse samples were obtained by instructing each subject to vigorously swish his/her mouth with 10 ml of sterile phosphate-buffered saline (PBS) for 1 min and then spit it out into a sterile container. Sampling was performed no less than 2 h after a meal. The samples were immediately transported to the laboratory and centrifuged at 3,500g for 3 min in sterile falcon tubes. The resultant pellets were each resuspended in 1 ml TE buffer and stored at −20°C.
DNA extraction
A DNA extraction protocol combining chemical lysis, glass bead-beating, and silica-column purification was used as follows. Samples were centrifuged at 10,000g for 2 min; the resultant pellets were each suspended in 360 µl of the digestion buffer, and 40 µl of the proteinase K included in the Purelink Genomic DNA extraction kit (Life Technologies, St. Louis, MO, USA); 0.2 g of sterile 0.5-mm glass beads (Sigma, Carlsbad, CA, USA) was added, and the mix was shaken for 3 min on a Mini-BeadBeater-8 (Biospec, Bartlesville, OK, USA) followed by incubation at 55°C for 2 h. Isolation of DNA was then performed following the Purelink's kit manufacturer instructions, using 100 µl of the supplied buffer for elution. The yield and quality of extracted DNA were checked using Nanodrop (Thermo Scientific, Waltham, MA, USA).
Amplicon library preparation and sequencing
Library preparation and sequencing were performed by GATC Biotech (Konstanz, Germany) as previously described (Citation23). In brief, the degenerate primers 27FYM (Citation24) and 519R (Citation25) were used to amplify the V1–V3 region of the 16S rRNA gene as described by Kistler et al. (Citation22), with a slight modification involving a separate PCR reaction with 2–3 cycles to incorporate the GS FLX titanium adaptors FLX-A and FLX-B as well as 5-base barcodes to the amplicons. The 12 tagged amplicon libraries were pooled with 8 other libraries (another study) in equimolar amounts and sequenced unidirectionally (side A) on quarter plate using 454 GS FLX chemistry (Roche, Germany).
Preprocessing of sequencing data
The raw data were submitted to Sequence Read Archive (SRA) under project accession number SRP049918. Data preprocessing was performed using the mothur software package version 1.33.3 (Citation26) as previously described with minor modification (Citation23). In brief, reads were filtered out and trimmed so as to minimize sequencing errors and include reads that were at least 380 bases long; reads with poor alignment were then removed and the rest were checked for chimeras with both UCHIME (Citation27) and ChimeraSlayer (Citation28), using the combined SILVA-HOMD set as reference.
Reference databases and reads classification algorithm
Three 16S rRNA gene reference sequence sets were used for taxonomic assignment of the reads: an updated version of the Human Oral Microbiome Database (updated-HOMD 13.2), a chimera-free version of the Human Oral Microbiome extended database (trusted-HOMDext), and a modified version of the Greengene Gold (modified-GGG) reference set (Citation23) – the fasta and taxonomy files for the three sets can be downloaded at ftp://www.homd.org/publication_data/20150519/.
The chimera-free reads were classified using the prioritized, multistage read assignment approach recently described (Citation23). In brief, the algorithm performed three stages of BLASTN (version 2.2.23) search to match reads at alignment coverage and percent identity of ≥98% to the reference sequences in the following order: the updated-HOMD 13.2, trusted-HOMDext, and modified-GGG (prioritization). At each stage, reads with single species top hit (a match with highest bit score and percent identity) were assigned to the corresponding unique species taxonomy whereas unmatched reads and those with multiple species top hits (of same score and percent identity) were searched against the next reference set for species-level identification with the same criteria. In the final stage, reads with multispecies best hits were annotated at the ‘species-group’ level (for consistent species combinations, e.g. Neisseria flavescens/subflava) or genus level (for inconsistent species combinations). Reads without hits in any of the three reference sets were subject to OTU analysis by clustering at 98% identity. Representative sequences of non-rare OTUs (those with more than three members) were BLASTN searched against NCBI's bacterial 16S rRNA sequences and nucleotide collection; OTUs returning hits were classified to the species level using the same criteria (both alignment length and identity ≥98% with highest score) whereas those unmatched were labeled as potential novel taxa, and classified to a higher rank (genus or family) using the Wang method (Citation29) and Greengene sequences as reference set.
The BLASTN search was performed on Linux-based computer servers provided by the HOMD team at the Forsyth Institute. The search results were parsed and best match(es) of each read was identified with a custom PERL script designed by the last author (CT) at Forsyth. The OTU calling for the unmatched reads and downstream analyses were done using Mothur on a personal computer by the first author (ANN).
Statistical analysis
The data were described at the phylum, genus, and species levels in terms of prevalence and relative abundance within and across the study subjects. A core bacteriome was defined as taxa present in at least 11 of the 12 subjects. The mothur software package version 1.33.3 was used to calculate the Chao 2, abundance-based coverage estimator (ACE), and Shannon index, and to generate rarefaction curves. The heatmap representing the core species was done using the ‘heatmap3’ function under the R statistical programming package version 3.0.1 (Citation30), and the hierarchical clustering was done with the ‘uclust’ function and the UPGMA method in R.
Results
Pyrosequencing information
In total, 138,000 raw reads were obtained (mean of 11,500 reads per sample). Around 30% of these were removed by applying the read quality and length filters. Additional 1,536 reads that did not align were excluded. UCHIME and Chimera Slayer identified 33.5% of the remaining reads as chimeric; removing these left behind 62,644 reads (mean of 5,220±781 reads/sample) with average length of 425 bases.
Taxonomic assignment of reads
Around 94% of the reads were matched to sequences in the three reference sets as follows: 91.8% to updated-HOMD 13.2, 1.2% to trusted-HOMDext, and 0.9% to modified-GGG. The vast majority of these reads (99%) were classified to the species level, either single species/phylotype (90%) or species-group level (9%). OTU analysis of the remaining 6% unmatched reads, resulted in 167 non-rare species-level OTUs, of which 113 OTUs representing 1,205 reads (additional 2%) were matched to reference NCBI sequences; the majority of these were human clones including 32 from the oral cavity. The remaining 54 OTUs were designated as potentially novel.
The core bacteriome – phylum and genus levels
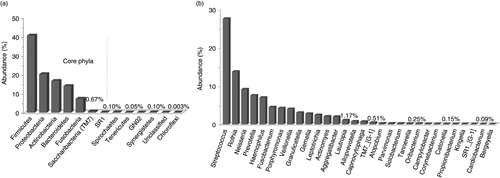
Overall, 13 phyla and 122 genera, all belonging to domain bacteria, were identified of which only 7 phyla and 30 genera fulfilled the definition of core taxa, that is, detected in at least 11 of the study subjects (). Spirochaetes, Tenericutes, Gracilibacteria (GN02), and Synergistetes were identified in nine, seven, six, and three subjects, respectively, whereas Chloroflexi and an unclassified phylum were seen in single samples. Forty-nine genera were found in at least six subjects and 80 in at least three subjects; 13 were unclassified. A list of all detected genera is provided in Supplementary Table 1. Five core phyla (Firmicutes, Proteobacteria, Actinobacteria, Bacteroidetes, and Fusobacteria) were present in high abundance, accounting for 98.8% of the sequences; the other two, Saccharibacteria (TM7) and SR1, were detected at very low abundance, together making up 0.77% of the sequences. The 30 core genera constituted 96.6% of the sequences; Streptococcus and Rothia were the most abundant (27.7 and 13.8% of the sequences, respectively) followed by Neisseria, Prevotella, Haemophilus, Fusobacterium, Porphyromonas and Veillonella constituting together nearly 80% of all reads.
Fig. 1. Distribution of phyla and genera across samples. (a) Thirteen phyla were detected, of which 7 were present in at least 11 of the 12 subjects (core phyla). (b) Relative abundance of 30 core genera out of 122 ones detected.
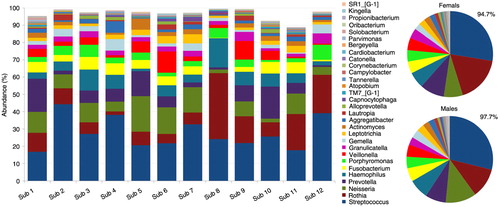
Distribution of the 13 phyla by subject and gender is shown in . The five high-abundance core phyla represented at least 96% of the reads in each of the study subjects. Compared to the males, the females harbored higher proportions of Actinobacteria, TM7, Spirochaetes, and Synergistetes at the expense of Proteobacteria. presents distribution of the 30 core genera by subject and gender. They constituted ≥90% of the bacteriome in each of the study subjects, averaging 94.7 and 97.7% in females and males, respectively. Ten of these (Streptococcus, Rothia, Neisseria, Prevotella, Haemophilus, Fusobacterium, Porphyromonas, Veillonella, Granulicatella, and Gemella) accounted for at least 74% of the sequences in each subject. The females tended to have higher proportions of Rothia, TM7 genus 1, Atopobium, Tannerella, Campylobacter, and Corynebacterium but lower proportions of Neisseria, Solobacterium, Propionibacterium, and Kingella.
Fig. 2. Distribution of phyla by subject and gender. Five core phyla (Firmicutes, Proteobacteria, Actinobacteria, Bacteroidetes and Fusobacteria) represented at least 96% of the reads. The females harbored higher proportions of Actinobacteria, TM7, Spirochaetes, and Synergistetes.
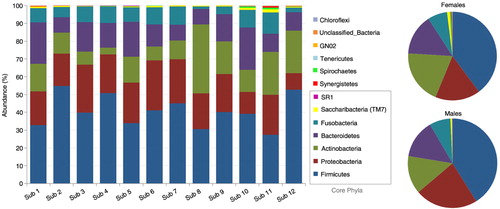
Fig. 3. Distribution of core genera by subject and gender. The thirty core genera constituted ≥89% of the microbiome in each of the study subjects, 94.7 and 97.7% in females and males, respectively. Ten of these (Streptococcus-Granulicatella in the figure key) accounted for at least 74% of the sequences.
The core bacteriome – species level
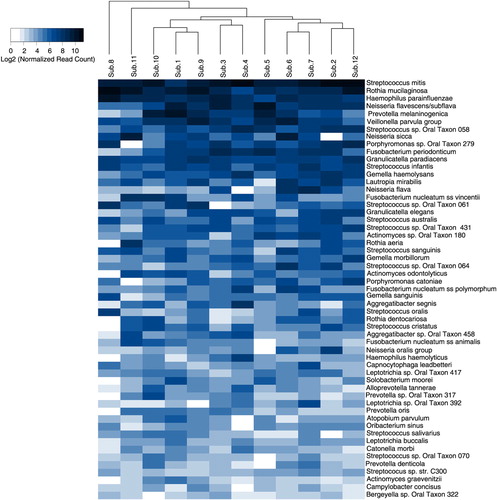
Five-hundred species/phylotypes, 3 species groups, and 54 potential novel OTUs (557 species-level taxa in total) were identified in the samples (Supplementary Table 2). Of these, 55 taxa constituted the core bacteriome, that is, they were present in at least 11 subjects (), whereas 347 taxa were detected in at least 3 subjects and 172 in at least 6 subjects. The core species made up between 67 and 87% of individual bacteriome, 80.7% on average. Overall, Streptococcus mitis, Rothia mucilaginosa, Haemophilus parainfluenzae, Neisseria flavescence/subflava group, Prevotella melaninogenica, and Veillonella parvula group were the most abundant, accounting in average for 15.28%, 12.53%, 6.13%, 4.94%, 3.97%, and 3.69% of the sequences, respectively.
Fig. 4. Heatmap of core species. The 55 core species (detected in at least 11 subjects) are ordered by their relative abundance across the study subjects. The map shows the relative abundance, normalized and log2-transformed, of the species within each subject. The subjects are clustered accordingly. Veillonella parvula group: V. parvula, V. dispar, V. atypica, and V. rogosae. Neisseria oralis group: N. oralis and Neisseria oral taxa 014 and 016.
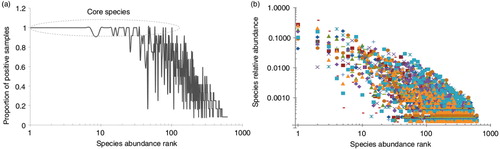
There was a significant correlation between species prevalence and abundance (Spearman's correlation coefficient=0.88), that is, the higher the prevalence, the higher the abundance was (A). For example, the core species represented the 25 most abundant, 39 out of the 50 most abundant and 52 out of the 100 most abundant taxa. However, a few taxa with low prevalence such as Fusobacteria sp. oral taxon A71, Leptotrichia sp. oral taxon B57, Eubacterium saphenum, Prevotella multiformis, and Aggregatibacter actinomycetemcomitans were detected at relatively high abundance. In addition, there were significant differences, up to three orders of magnitude, in the relative abundance of individual species, including core ones, among the study subjects (B).
Fig. 5. Species prevalence and relative abundance. The species rank is defined by its average abundance across all samples, with the most abundant species ranked first (left). (a) Species prevalence, the proportion of samples containing that species, plotted against species rank. (b) The relative abundance plotted for each sample in which the species is present – that is, for each species rank, a mark represents a sample positive for that species.
Species diversity and richness
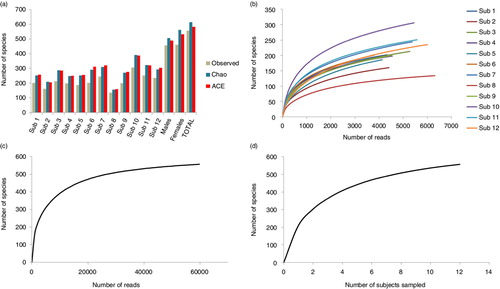
A mean of 211±42 species-level taxa was observed per subject (range 134–306). The mean estimated richness, calculated by Chao2 and ACE, was 273±56 and 278±56 species-level taxa per subject, respectively, that is, around 30% higher than the observed richness (A). Taking all samples together, however, the difference between the observed and estimated richness was much lower (586 and 614 species-level taxa by ACE and Chao2, respectively, compared to the 557 observed). The Shannon index ranged from 2.5 to 4.1 for individual samples, averaging 3.99 across samples. The rarefaction curves (B–D) show that increasing the number of subjects sampled or depth of sequencing within, but not across, subjects would have allowed detection of additional species. Species coverage ranged between 98.3 and 99.5% for individual samples; 99.9% for all samples combined.
Fig. 6. Species richness and rarefaction curves. (a) Observed and estimated species richness within each subject, in the males and females and across subjects. ACE: abundance-based coverage estimator. The rarefaction curves were generated by plotting the number of species as a function of (b) number of reads from each subject, (c) number of total reads, and (d) number of sampled subjects.
Discussion
This study, to the best of our knowledge, is the first description of the oral bacteriome in an Arab population. The study is also the first attempt to characterize the core oral bacteriome at the strict species level based on NGS reads data. Stringent selection criteria of participants were used as detailed in the materials and methods section to minimize variations in the composition of the oral bacteriome. However, the sample size was somewhat small, probably resulting in underestimation of species richness and diversity, which presents one study limitation.
Vigorous swishing was used for sampling on the assumption that it would improve species recovery from dental sites and thus the samples’ representativeness of the oral bacteriome. Nevertheless, subgingival species were probably underrepresented which is another study limitation. The DNA extraction protocol included a bead-beating step because it has been shown to improve species recovery (Citation16). The samples were sequenced at an initial depth of ~11,500 reads per sample; however, stringent quality filtration and chimera removal were applied to ensure reliable classification of the reads, which reduced the sequencing depth by almost 50%. The remaining reads were classified using the prioritized multistage BLASTN-based classification algorithm recently described (Citation23). By using multiple reference data sets, the algorithm maximizes the fraction of reads classified at the species level while ensuring reliable classification by giving priority to the human oral reference set. Although computationally demanding – because of the slow BLASTN search of individual reads – the gain in species-level assigning accuracy outweighs the loss in speed, which is in fact less of an issue nowadays with the availability of affordable multi-core computation platforms.
In total, 557 species-level taxa were identified with an average of 211 taxa per subject. In comparison, the number of species-level OTUs identified in previous studies ranged between as low as 247 and as high as 5,592 OTUs (Citation7, Citation8) (Citation13, Citation15) (Citation17, Citation22). This significant variation may be attributed to differences in methodological aspects including selection of the 16S rRNA hypervariable region, read quality control, type of sample, number of subjects included and, most importantly, OTU calling and taxonomy assigning methods. Specifically, it seems that de novo clustering of reads into OTUs significantly inflates species richness. For example, reads in the current study that successfully matched sequences in one of the three reference data sets (i.e. excluding unassigned reads that were later subject to de novo OTU calling) were assigned to 413 species-level taxa. Attempting to cluster these reads at 98% percent identity using average neighborhood resulted in 4,504 OTUs, the majority of which were singletons (data not shown). Diaz et al. (Citation16) encountered similar findings using a mock community; however, they attributed them to spurious OTUs resulting from sequencing errors and suggested eliminating singletons to overcome the problem. Our analysis, on the other hand, demonstrates that singletons are frequently high quality reads that can be reliably classified, and that eliminating them is not justified. A better explanation of singletons accordingly can be that de novo clustering undesirably bins reads representing single species into multiple OTUs. In fact, de novo OTU calling can also bin reads of multiple species into a single OTU (especially true for close species with 16S rRNA sequence similarity higher than the percent identify cutoff used in OTU calling). This argument highlights further the limitations of de novo OTU calling and the advantage of the reference-based reads classification algorithm used in this study.
The concept of core oral bacteriome (or microbiome) is still evolving. A number of recent studies described the core oral bacteriome in health (Citation7, Citation10) (Citation15, Citation16) (Citation18, Citation21) (Citation22, Citation31) but have not been consistent with respect to its definition, and only a few of them reported species-level taxonomic assignments for the shared OTUs (Citation16, Citation21) (Citation22). In this study, the core bacteriome comprised 55 species-level taxa. Depending on the thresholds used for clustering the reads into OTUs and defining the core bacteriome, the number of core OTUs found in previous studies ranged between 3 (Citation10) and 504 (Citation15); an extreme of 3,530 OTUs has also been reported (Citation32). Given the inherent limitations of OTU analysis mentioned above, interpretation of and comparisons with these numbers should be done with caution. In fact, when studies that assigned species-level taxonomy to OTUs were considered separately, we found that the highest number of core species reported was 78, which is comparable to the number reported in this study and suggests that a higher taxonomic resolution probably results in better characterization of the core bacteriome.
With the exception of Streptococcus sp. C300, all of the core species identified in the current study were human oral taxa with reference sequences in updated-HOMD 13.2. The former is a fully sequenced human respiratory isolate, which has never been described before as member of the oral microbiota; failure of previous studies to identify it in oral samples is probably because it has been described only quite recently (2013) and a representative 16S rRNA of it had thus not been included in the reference sets used for classification. S. mitis was the most abundant core species on average, which is consistent with previous reports of the normal oral bacteriome (Citation16, Citation22) (Citation33). The second most abundant core species was R. mucilaginosa. This species is a common colonizer of the tongue (Citation34) and has been identified in one study as an abundant core species of the salivary bacteriome in healthy subjects (Citation16), though not as abundant as shown in the current study. However, another Rothia species, namely, R. dentocariosa, has been more frequently reported as core and abundant member of the oral bacterial community in health (Citation21, Citation22). Although this species was also identified as a core species in the current study, it was not abundant. H. parainfluenzae ranked as the third most abundant core species, which is consistent with several previous reports in which this taxon has been identified as a core or/and abundant species in association with health (Citation7, Citation10) (Citation16, Citation22).
Of the other core species identified, V. parvula, N. flavescence/subflava, P. melaninogenica, Fusobacterium periodonticum, Fusobacterium nucleatum ss polymorphum, Granulicatella adiacens, Gemella haemolysans, Lautropia mirabilis, Granulicatella elegan, Rothia aeria, Actinomyces odontolyticus, Porphyromonas catoniae, Streptococcus sanguinis, and Neisseria sicca have also been described elsewhere as being core species in association with health (Citation7, Citation16) (Citation21, Citation22). So, there is increasing evidence for these species as members of a ‘universal’ core human oral bacteriome. The remaining, low-abundance core species may be ethnicity-specific, which remains to be investigated further. What is clear, however, is that there are significant differences in the abundance of core species among individuals, accounting for the interindividual variations reported in previous studies (Citation10, Citation11) (Citation14).
More consistency is found among studies at the genus and phylum levels. In this study, 122 genera were identified, which falls within the range of 51–135 genera reported in the literature (Citation6–Citation8, Citation11) (Citation13, Citation15) (Citation18, Citation22) (Citation31), excluding the study by Keijser et al. (Citation17), which is an extreme outlier at all taxonomy levels. Thirty genera were found to constitute the core bacteriome, many of which, including Streptococcus, Rothia, Neisseria, Prevotella, Veillonella, Haemophilus, Fusobacterium, Porphyromonas, Granulicatella, Gemella, Leptotrichia, Actinomyces, Aggregatibacter, Lautropia, and Capnocytophaga, have been repeatedly described among the most common and/or abundant genera in oral samples (Citation7, Citation9) (Citation11–Citation16, Citation31). Interestingly, SR1 [G-1] is for the first time reported as a core genus, which may be specific to the Arab population. Consistent with findings from this study, the number of phyla identified in the majority of previous studies has been in the range of 9–12 (Citation6–(Citation9), Citation 13 (Citation15, Citation22). Firmicutes, Proteobacteria, Actinobacteria, Bacteroidetes, and Fusobacteria are unanimously described as the most abundant phyla in the oral cavity and thus represent a universal core. In the current study, SR-1 and TM7 were also identified as core phyla, albeit with lower abundance. The latter has been described as a core phylum by Lazarevic et al. (Citation31); SR-1 has also been identified in other studies but whether or not it was present in all samples was not mentioned (Citation6, Citation8) (Citation9).
In conclusion, the present study describes a high resolution, species-level, core oral bacteriome based on NGS data. The core species accounted for the majority of reads in the samples; that is, they were the most abundant. Comparison with previous studies suggests that some of these species may represent ‘universal’ members of the core oral human bacteriome, whereas others may be ethnicity-specific. Whether there is a true universal healthy human core oral bacteriome, ethnicity-specific species members require a larger-scale study with standard experimental protocol and reads assignment method.
Conflict of interest and funding
The authors declare that they have no conflict of interests.
High resolution figures + supplementary results
Download (3.8 MB)Acknowledgements
The study was funded by SABIC through the Deanship of Scientific Research at Jazan University, Saudi Arabia (grant #33/4/35). We thank Dr. Mohammed Muzaffer Ali Khan, Dr. Ahmed Alqahtani, and Dr. Yousef Al-shamrani for their kind help with collection of the samples.
Notes
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
Related Research Data
References
- Chen H, Jiang W. Application of high-throughput sequencing in understanding human oral microbiome related with health and disease. Front Microbiol. 2014; 5: 508. [PubMed Abstract] [PubMed CentralFull Text].
- Sato Y, Yamagishi J, Yamashita R, Shinozaki N, Ye B, Yamada T, etal. Inter-individual differences in the oral bacteriome are greater than intra-day fluctuations in individuals. PLoS One. 2015; 10: e0131607. [PubMed Abstract] [PubMed CentralFull Text].
- Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, etal. The human oral microbiome. J Bacteriol. 2010; 192: 5002–17. [PubMed Abstract] [PubMed CentralFull Text].
- Marsh PD. Are dental diseases examples of ecological catastrophes?. Microbiology. 2003; 149: 279–94. [PubMed Abstract].
- Siqueira JF Jr., Fouad AF, Rocas IN. Pyrosequencing as a tool for better understanding of human microbiomes. J Oral Microbiol. 2012; 4: 10743. doi: http://dx.doi.org/10.3402/jom.v4i0.10743.
- Ahn J, Yang L, Paster BJ, Ganly I, Morris L, Pei Z, etal. Oral microbiome profiles: 16S rRNA pyrosequencing and microarray assay comparison. PLoS One. 2011; 6: e22788. [PubMed Abstract] [PubMed CentralFull Text].
- Bik EM, Long CD, Armitage GC, Loomer P, Emerson J, Mongodin EF, etal. Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J. 2010; 4: 962–74. [PubMed Abstract] [PubMed CentralFull Text].
- Contreras M, Costello EK, Hidalgo G, Magris M, Knight R, Dominguez-Bello MG. The bacterial microbiota in the oral mucosa of rural Amerindians. Microbiology. 2010; 156: 3282–7. [PubMed Abstract].
- Huang S, Yang F, Zeng X, Chen J, Li R, Wen T, etal. Preliminary characterization of the oral microbiota of Chinese adults with and without gingivitis. BMC Oral Health. 2011; 11: 33. [PubMed Abstract] [PubMed CentralFull Text].
- Huse SM, Ye Y, Zhou Y, Fodor AA. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One. 2012; 7: e34242. [PubMed Abstract] [PubMed CentralFull Text].
- Nasidze I, Li J, Quinque D, Tang K, Stoneking M. Global diversity in the human salivary microbiome. Genome Res. 2009; 19: 636–43. [PubMed Abstract] [PubMed CentralFull Text].
- Simon-Soro A, Tomas I, Cabrera-Rubio R, Catalan MD, Nyvad B, Mira A. Microbial geography of the oral cavity. J Dent Res. 2013; 92: 616–21. [PubMed Abstract].
- Zaura E, Keijser BJ, Huse SM, Crielaard W. Defining the healthy ‘core microbiome’ of oral microbial communities. BMC Microbiol. 2009; 9: 259. [PubMed Abstract] [PubMed CentralFull Text].
- Li J, Quinque D, Horz HP, Li M, Rzhetskaya M, Raff JA, etal. Comparative analysis of the human saliva microbiome from different climate zones: Alaska, Germany, and Africa. BMC Microbiol. 2014; 14: 316. [PubMed Abstract] [PubMed CentralFull Text].
- Ling Z, Liu X, Luo Y, Yuan L, Nelson KE, Wang Y, etal. Pyrosequencing analysis of the human microbiota of healthy Chinese undergraduates. BMC Genomics. 2013; 14: 390. [PubMed Abstract] [PubMed CentralFull Text].
- Diaz PI, Dupuy AK, Abusleme L, Reese B, Obergfell C, Choquette L, etal. Using high throughput sequencing to explore the biodiversity in oral bacterial communities. Mol Oral Microbiol. 2012; 27: 182–201. [PubMed Abstract] [PubMed CentralFull Text].
- Keijser BJ, Zaura E, Huse SM, van der Vossen JM, Schuren FH, Montijn RC, etal. Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res. 2008; 87: 1016–20. [PubMed Abstract].
- Mason MR, Nagaraja HN, Camerlengo T, Joshi V, Kumar PS. Deep sequencing identifies ethnicity-specific bacterial signatures in the oral microbiome. PLoS One. 2013; 8: e77287. [PubMed Abstract] [PubMed CentralFull Text].
- Schloss PD, Westcott SL. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl Environ Microbiol. 2011; 77: 3219–26. [PubMed Abstract] [PubMed CentralFull Text].
- Chen T, Yu WH, Izard J, Baranova OV, Lakshmanan A, Dewhirst FE. The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford) 2010. 2010. baq013.
- Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, etal. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013; 7: 1016–25. [PubMed Abstract] [PubMed CentralFull Text].
- Kistler JO, Booth V, Bradshaw DJ, Wade WG. Bacterial community development in experimental gingivitis. PLoS One. 2013; 8: e71227. [PubMed Abstract] [PubMed CentralFull Text].
- Al-Hebshi NN, Nasher AT, Idris AM, Chen T. Robust species taxonomy assignment algorithm for 16S rRNA NGS reads: application to oral carcinoma samples. J Oral Microbiol. 2015; 7: 28934. [PubMed Abstract].
- Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, Olsen GJ. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl Environ Microbiol. 2008; 74: 2461–70. [PubMed Abstract] [PubMed CentralFull Text].
- Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci USA. 1985; 82: 6955–9. [PubMed Abstract] [PubMed CentralFull Text].
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, etal. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009; 75: 7537–41. [PubMed Abstract] [PubMed CentralFull Text].
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011; 27: 2194–200. [PubMed Abstract] [PubMed CentralFull Text].
- Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, etal. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011; 21: 494–504. [PubMed Abstract] [PubMed CentralFull Text].
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007; 73: 5261–7. [PubMed Abstract] [PubMed CentralFull Text].
- R Core Team. R: A language and environment for statistical computing . 2013; Vienna, Austria: R Foundation for Statistical Computing.
- Lazarevic V, Whiteson K, Hernandez D, Francois P, Schrenzel J. Study of inter- and intra-individual variations in the salivary microbiota. BMC Genomics. 2010; 11: 523. [PubMed Abstract] [PubMed CentralFull Text].
- Jiang W, Ling Z, Lin X, Chen Y, Zhang J, Yu J, etal. Pyrosequencing analysis of oral microbiota shifting in various caries states in childhood. Microb Ecol. 2014; 67: 962–9. [PubMed Abstract].
- Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005; 43: 5721–32. [PubMed Abstract] [PubMed CentralFull Text].
- Kazor CE, Mitchell PM, Lee AM, Stokes LN, Loesche WJ, Dewhirst FE, etal. Diversity of bacterial populations on the tongue dorsa of patients with halitosis and healthy patients. J Clin Microbiol. 2003; 41: 558–63. [PubMed Abstract] [PubMed CentralFull Text].