
Abstract
The aim of the study was to determine the multilocus sequence types of Escherichia coli from diseased farm rabbits and apparently healthy wild lagomorphs, and the genetic relatedness among them. Fifty-five enteropathogenic E. coli from reared rabbits and 32 from wild rabbits and hares were characterised by multilocus sequence typing (MLST) according to the Michigan State University Ec MLST scheme. Isolates were differentiated into 37 sequence types (STs), which were grouped into 8 clonal complexes (CCs). The most common ST was ST140 (CC31), followed by ST238 and ST119 (CC17). MLST analysis revealed 22 novel STs. Phylogenetic analyses showed a heterogeneous distribution of STs into 3 clusters of genetically related strains. The genetic relationship among STs of different origin and the detection of new, as well as previously described STs as human pathogens, indicate a widespread distribution and adaptability of particular lineages to different hosts. These findings highlight the need for further research to improve the knowledge about E. coli populations colonising the gut of lagomorphs and their zoonotic potential.
Introduction
Escherichia coli are commensal bacteria colonising the intestinal tract of a wide range of animals and humans. However, some adapted clones are potentially pathogenic. These clones are considered to be among the major causes of gastrointestinal disease in humans and are responsible for economic losses in the animal farm industry worldwide (Dezfulian et al., Citation2003; Kaper et al., Citation2004).
Rabbits (Oryctolagus cuniculus) are one of the most reared animals for meat production in several countries; Italy is the leader country within Europe (FAO, Citation2014). These animals are highly susceptible to diseases, and enteric infections (especially caused by E. coli) represent the most serious health problem in rabbit farms (Blanco et al., Citation1996; Boullier and Milon, Citation2006; Agnoletti, Citation2012). Although E. coli naturally colonises the guts of reared and wild lagomorphs, rabbit enteropathogenic E. coli strains (EPEC) are responsible for significant economic losses in the rabbit meat industry, mainly due to decreased growth, high mortality rates and drug-related costs (Boullier and Milon, Citation2006).
Although the pathogenic strains circulating in rabbit farms are a source of concern due to their economic impact and their potential public health implications, E. coli colonisation of wild species is of interest as well. In addition to the key ecological function affecting free-living animals in different niches, wild rabbits (Oryctologus cuniculus) may also play a role as reservoirs of potentially zoonotic microorganisms and therefore should be monitored (Scaife et al., Citation2006; Silva et al., Citation2010). European wild rabbits and hares (Lepus europaeus europaeus) have great ecological and economic value since they are key species in Mediterranean ecosystems, important game animal for sport hunting and are destined for human consumption, despite the presence of other free-living species (Delibes et al., Citation2008).
To date, the genetic diversity of EPEC infecting rabbits remains undefined. Moreover, little is known about the genetic relationships and host specificities among isolates from different lagomorph populations. Specifically, it is not clear whether particular genotypes exhibit different abilities to colonise these animal species. Over the last decade, the implementation of molecular biology techniques has provided useful methods, such as multilocus sequence typing (MLST), to compare strains. Recently, several authors have reported the use of molecular typing methods to study E. coli and have provided evidence for the crucial role of some sequence types (STs) or clonal complexes (CCs) in causing infection. Furthermore, studies have demonstrated possible routes of transmission between animals and humans (Bidet et al., Citation2007; Moura et al., Citation2009; Platell et al., Citation2011).
To the best of our knowledge no surveys have been performed to determine the molecular epidemiology of E. coli isolated from farm rabbits and wild lagomorphs in Italy. Indeed, reports characterising E. coli from these animal species are limited and usually restricted to the analysis of sero/biotypes, antimicrobial-resistance profiles and pathogenicity (Agnoletti et al., Citation2004; Hassan and Al-Azeen, Citation2009; Poeta et al., Citation2010; Badagliacca et al., Citation2012; Marinho et al., Citation2014).
Based on these considerations, the aims of the present work were: i) to determine the multilocus sequence types of E. coli circulating in diseased farm rabbits and apparently healthy wild lagomorphs; and ii) to clarify the phylogenetic relatedness among genotypes.
Materials and methods
E. coli strains analysed in this study were isolated from the intestinal contents of reared rabbits (O. cuniculus), wild rabbits (O. cuniculus) and hares (Lepus europaeus europaeus) monitored or hunted in northern Italy between 2006 and 2008.
All strains were isolated using classical microbiological standard procedures and were previously characterised by Dotto et al. (Citation2014). As shown in , further 16 E. coli strains collected during the same period were added to the 71 isolates previously characterised. All 55 clinical strains isolated from unrelated outbreaks occurred in different Italian commercial rabbit farms were categorised as EPEC for the presence of the eae gene following the procedure described by Penteado et al. (Citation2002), whereas the 32 isolates from wild lagomorphs were classified as commensals. All commensal isolates were collected from apparently healthy wild lagomorphs (15 strains from wild rabbits and 17 from hares) hunted during the shooting season or tested during veterinary periodical monitoring programs to evaluate the clinical conditions of free-living animals.
Table 1. Animal species, year of isolation, geographical origin, allelic profiles, sequence types and clonal complexes of Escherichia coli isolated from farm rabbits, wild rabbits and hares sampled in Lombardy, Veneto and Piedmont in the period between 2006-2008.
Isolates were revitalised on nutrient agar (OXOID, Basingstoke, UK), incubated aerobically overnight at 37°C, and processed for the isolation of genomic DNA. Briefly, the cultures were suspended in sterile water and boiled for 10 min. The supernatants containing DNA were used as templates for molecular assays.
The strains were analysed by MLST as described by the Michigan State University Ec MLST scheme (http://www.shigatox.net/ecmlst/cgibin/index). PCR was performed using the primers and the protocols specified on the website. Amplicons were purified using the PCR Cleanup Kit (Macherey-Nagel, Düren, Germany) and both strands were sequenced using the BigDye Terminator Cycle Sequencing Kit v3.1 (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s instructions. The reaction products were separated and detected by capillary electrophoresis using the ABI PRISM 3100 Genetic Analyser (Applied Biosystems).
Raw nucleotide sequences were analysed and assembled using ChromasPro v1.42 (Technelysium Pty Ltd, Tewantin, Australia). The allelic profiles, STs and CCs were obtained by submitting the allele sequences to the Ec MLST web site. Allele sequences and profiles, not previously reported, were assigned with new allele numbers and STs by the curator of the database. Each isolate was assigned a sequence type (ST) according to its allelic profile.
The sequences from all 7 loci of each bacterial strain were concatenated for phylogenetic analysis. Then, the concatenated sequences were aligned using the MAFFT algorithm of the Guidance software (http://guidance.tau.ac.il/ver2/). The phylogenetic trees were constructed using the Maximum Likelihood algorithm with the RAxML 7.3.0 software, and branch support was evaluated by performing 1000 bootstrap replicates (Stamatakis, Citation2006). To understand the epidemiological links and the phylogenetic relationships among STs, networks were constructed using the goeBURST algorithm in the PHYLOVIZ software (http://goeburst.phyloviz.net/). Strains of different STs sharing at least 5 of the 7 alleles were interpreted as belonging to the same clonal complex (CC).
Results and discussion
E. coli isolates were differentiated into 37 different STs and grouped into 8 CCs (). Out of 37 different STs (21 from farm rabbits, 8 from wild rabbits and 14 from hares), 16 were represented by more than 1 isolate, whereas 21 were each represented by 1 strain. Only 15 of the different profiles could be retrieved from the MLST database, while 22 were new (ST1004 to ST1025) (). Although most of the new STs were the result of novel combinations of previously described alleles, 5 (i.e. ST1018 to ST1021 and ST1025) were new alleles differing by 1 nucleotide from their respective counterparts in the nearest STs. Of the 15 known STs (), ST140 was the most commonly detected (8 from farm rabbits and 2 from hares), followed by ST238 (8 from farm rabbits and 1 from a wild rabbit), ST119 (5 from farm rabbits and 3 from wild rabbits), ST148 (3 from farm rabbits and 1 from a wild rabbit) and ST259 and ST461 with 2 strains each (ST259 from 1 farm rabbit and 1 hare; ST461 from 2 farm rabbits). The other 9 known STs were represented by 1 isolate and were equally distributed among farm rabbits and hares. The most common new allelic profile (ST1014) was represented by 6 strains isolated from wild rabbits, followed by ST1007 and ST1006 (5 from farm rabbits and 4 and 3 from farm rabbits, and 1 from a hare), ST1008 and ST1019 (3 from hares and farm rabbits, respectively) and ST1004, ST1011, ST1012, ST1018 and ST1021 with 2 strains each, all isolated from farmed rabbits. Conversely, the remaining 12 new STs (i.e. ST1009, ST1010, ST1013, ST1015-ST1016 and ST1022-ST1025 from wild animals and ST1005, ST1017 and ST1020 from farm rabbits) were represented by only 1 isolate.
Novel STs were detected both in reared and wild lagomorphs. Interestingly, in contrast with the results described for the most prevalent profiles, most of the new STs represented by more than 1 isolate seemed to be species-specific (farm/wild rabbits or hares). Indeed, except for ST1006 recognised in isolates from both farm and wild rabbits, the other novel STs were described in either reared or wild lagomorphs. Therefore, novel STs may be genotypes adapted to particular lagomorph populations. However, it cannot be excluded that the high number of novel STs may be in part due to the overall paucity of data available from lagomorphs and to the collection of E. coli from a previously unsampled geographic area. Conversely, the predominant known STs seemed to be associated with widely diffused strains, especially (but not only) those isolated from industrial farms. Therefore, the widespread nature of the same STs as part of the pathogenic and commensal population makes difficult to determine whether there is any association of these STs with infection in farm rabbits or colonisation in wild lagomorphs.
Overall, the MLST analysis revealed a high genetic heterogeneity. This diversity is indicative of the complexity of the lagomorph enteric population. These data are in accordance with previous studies performed on isolates of human and poultry origin, suggesting that a broad variety of E. coli types are able to colonise the animal gut as commensals or cause disease in the same animal species (Jaureguy et al., Citation2008; Dissanayake et al., Citation2014). The high level of E. coli genetic diversity in isolates from free-living animals in combination with the high ratio of novel STs may be explained by the fact that these strains were isolated from randomly captured animals.
Although we observed diversity among isolates, most belonged to 2 main clonal groups (CC17 and CC31), indicating that these strains tend to be phylogenetically related (, ). Eight CCs were identified: CC17, CC31, CC63, CC41 and CC 30, CC7, CC23 and CC36. Overall, 30% of the isolates belonged to CC17 (17/87; 19.5% from farm and wild rabbits) and CC31 (11/87; 12.6% from farm rabbits and hares); however, most of the STs (26/37; 70.3%), and especially the novel profiles, were not included into a CC. The identified CCs are quite frequent among isolates of both animal and human origin. Indeed, although only a few studies are available, an analysis of the data from strains already reported in the Ec MLST database (accessed June 2015) or included in previous studies indicates that most of the isolates belonging to CC17 and CC31 had a human or a livestock origin (mainly cattle). Only 2 STs (ST119 and ST238) were previously isolated from rabbits, and most of them were pathogens or related to clinical conditions (Steinsland et al., Citation2010; Tennant et al., Citation2009). Strains from other animal species were less prevalent either in the Ec MLST database and in literature, and only few STs isolated from diarrhoeic rabbits were described in other studies (Moura et al., Citation2009; Steinsland et al., Citation2010). Despite fragmentary, information from the database suggests that most of the isolates with previously known STs (especially those belonging to CC17 and CC31) are globally widespread and able of colonising a wide range of animal species, including humans. In accordance with other studies, the identification of closely related clones residing in different hosts suggests that they might have no or minimal host specificity (Moura et al., Citation2009; Clermont et al., Citation2011). With the exception of novel profiles, the limited data deposited in the Ec MLST database on E. coli from rabbits makes difficult to determine the spread of already known STs in farm rabbits or their role as commensals in wild animals globally. However, these findings suggest a potential role of lagomorphs, mainly reared rabbits, as reservoirs of zoonotic bacteria. However, further studies should be conducted to ascertain this hypothesis. The phylogenetic tree of the 87 E. coli isolates constructed with the concatenated sequences of the 7 conserved housekeeping genes is shown in . The phylogenetic analysis grouped the strains into 3 main clusters (A, B and C). Most of the isolates (62/87; 71.3%) were grouped into cluster C, whereas the remaining 25 isolates grouped into clusters A (16/87; 18.4%) and B (9/87; 10.4%). Cluster A included strains isolated from farm rabbits (n=9) and hares (n=7), whereas none was from wild rabbits. The smallest group (cluster B) comprised only 9 strains, most of which (6/9 strains) isolated from wild rabbits (ST1014). Out of the 3 remaining strains, 2 were isolated from reared rabbits and 1 from a hare. Forty-four EPEC isolated from diseased farm rabbits were grouped into cluster C, especially those belonging to CC31, the closely related isolates of CC17 (ST119 and ST238) and the new ST1007. Altogether, these strains formed the most representative sub-group of cluster C. However, the same cluster included also strains from free-living animals, which shared mostly the same STs as the reared rabbits described above.
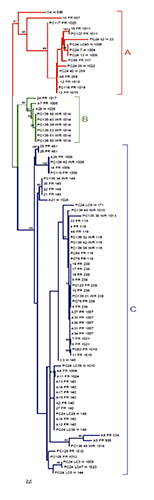
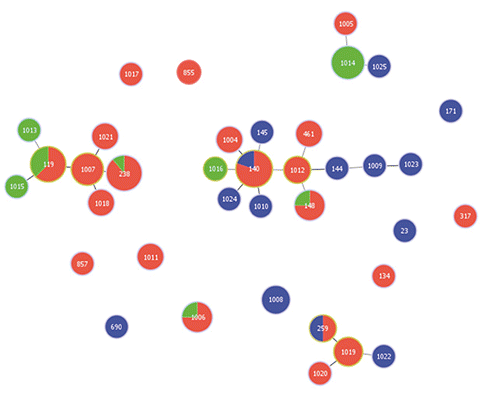
The heterogeneity of isolates was confirmed by the goeBURST analysis (), according to the constructed phylogenetic tree. These analyses were performed to identify CCs, which are groups of closely related genotypes sharing 5 loci with at least one other ST and probably descending from a common ancestor. As shown in , most of the strains belonged to 2 relevant CCs (CC17 and CC31), including a grouping of the most common STs (ST119, ST238, and ST140). Interestingly, some novel STs may have a common origin with those assigned to the prevalent CCs, as shown by the phylogenetic tree (). As shown in and , some strains isolated from healthy wild animals represented the same strains (i.e. ST119, ST140, ST148, ST238 and ST259) or revealed a close relationship with EPEC from reared animals. Based on these data, these strains and pathotypes may have evolved via either the loss or acquisition of virulence factors, but these events cannot be detected by MLST analysis of small fragments of conserved genes (Moura et al., Citation2009) and therefore further studies are necessary. In accordance with other studies, it is not unusual to find genetic diversity within strains of a similar ST, mainly due to rearrangements, recombination events or point mutations occurring along the genome and common in E. coli (Hao et al., Citation2012; Croxen et al., Citation2013). Indeed, it is well established that the acquisition of new genes, together with gene loss, may favour the adaptability of pathogens in particular hosts (Croxen et al., Citation2013).
Conclusions
This is the first study reporting the multilocus sequence types of E. coli isolated from domestic and wild lagomorphs, and new host-adapted strains together with widespread lineages were identified. This finding is of great value since it confirms the high adaptability of E. coli to various niches and its ability to cross the host barriers. However, further studies are needed to improve the knowledge about E. coli populations colonising the domestic and wild lagomorphs and their zoonotic potential.
Acknowledgements
This work was financed by grants from the Department of Comparative Biomedicine and Food Science, University of Padua and from the University of Padua (CPDA095771/09). The Authors wish to thank Dr. Hans Steinsland (Michigan State University, East Lansing, MI, USA) for his contribution to the www.shigatox.net/ecmlst website.
References
- AgnolettiF., 2012. Update on rabbit enteric diseases: despite improved diagnostic capacity, where does disease control and prevention stand? pp 1113–1127 in Proc. 10th World Rabbit Congr., Sharm El-Sheikh, Egypt.
- AgnolettiF. Favretti M. Deotto S. Passera A. Tisato E. Bano L. Mazzolini E., 2004. Report of enteropathogenic Escherichia coli (EPEC) isolated from enteric outbreaks in Italian intensive rabbit herds. pp 416–421 in Proc. 8th World Rabbit Congr., Puebla, Mexico.
- BadagliaccaP. Agnoletti F. Tonelli A. Masson L., 2012. Antimicrobial resistance-related gene profiles of rabbit enteropathogenic Escherichia coli strains isolated from colibacillosis outbreaks in Northern Italy. pp 1191–1195 in Proc. 10th World Rabbit Congr., Sharm El- Sheikh, Egypt.
- BidetP. Mahjoub-Messai F. Blanco J. Blanco J. Dehem M. Aujard Y. Bingen E. Bonacorsi S., 2007. Combined multilocus sequence typing and O serogrouping distinguishes Escherichia coli subtypes associated with infant urosepsis and/or meningitis. J. Infect. Dis. 196:297–303.
- BlancoJ.E. Blanco M. Blanco J. Mora A. Balaguer L. Mourini M. Juarez A. Jansen W.H., 1996. O serogroups, bio-types, and eae genes in Escherichia coli strains isolated from diarrheic and healthy rabbits. J. Clin. Microbiol. 34:3101–3107.
- BoullierS. Milon A., 2006. Rabbit colibacillosis. In: MaertensL. CoudertP. (eds) Recent advances in rabbit sciences. ILVO, Melle, Belgium, pp 171–179.
- ClermontO. Olier M. Hoede C. Diancourt L. Brisse S. Keroudean M. Glodt J. Picard B. Oswald E. Denamur E., 2011. Animal and human pathogenic Escherichia coli strains share common genetic backgrounds. Infect. Genet. Evol. 11:654–662.
- CroxenM.A. Law R.J. Scholz R. Keeney K.M. Wlodarska M. Finlay B.B., 2013. Recent advances in understanding enteric pathogenic Escherichia coli. Clin. Microbiol. Rev. 26:822–880.
- Delibes M. Delibes M. Ferreras P. Villafuerte R., 2008. Key role of European rabbits in the conservation of the Western Mediterranean basin hotspot. Conserv. Biol. 22:1106–1117.
- Dezfulian H. Batisson I. Fairbrother J.M. LauP.C. NassarA. Szatmari G. Harel J., 2003. Presence and characterization of extraintestinal pathogenic Escherichia coli virulence genes in F165-positive E. coli strains isolated from diseased calves and pigs. J. Clin. Microbiol. 41:1375–1385.
- Dissanayake D.R. Octavia S. Lan R., 2014. Population structure and virulence content of avian pathogenic Escherichia coli isolated from outbreaks in Sri Lanka. Vet. Microbiol. 168:403–412.
- DottoG. Grilli G. Giacomelli M. Ferrazzi V. Carattoli A. Fortini D. Piccirillo A., 2014. High prevalence of oqxAB in Escherichia coli isolates from domestic and wild lagomorphs in Italy. Microb. Drug Resist. 20:118–123.
- FAO, 2014. World production of rabbit meat in 2012. Available from: http://faostat.fao.org/site/291/default.aspx
- HaoW. Allen V.G. Jamieson F.B. Low D.E. Alexander D.C., 2012. Phylogenetic incongruence in E. coli O104: understanding the evolutionary relationships of emerging pathogens in the face of homologous recombination. PLoS One 7:e33971.
- HassanS.A. Al-Azeen M.W.A., 2009. Determination of virulence gene markers and antimicrobial resistance in Escherichia coli isolated from rabbit in Egypt. Global Veterinaria 3:260–267.
- JaureguyF. Landraud L. Passet V. Diancourt L. Frapy E. Guigon G. Carbonnelle E. Lortholary O. Clermont O. Denamur E. Picard B. Nassif X. Brisse S., 2008. Phylogenetic and genomic diversity of human bacteremic Escherichia coli strains. BMC Genomics 9:560.
- KaperJ.B. Nataro J.P. Mobley H.L., 2004. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2:123–140.
- MarinhoC. Igrejas G. Gonçalves A. Silva N. Santos T. Monteiro R. Gonçalves D. Rodrigues T. Poeta P., 2014. Azorean wild rabbits as reservoirs of antimicrobial resistant Escherichia coli. Anaerobe 30:116–119.
- MouraR.A. Sircili M.P. Leomil L. Matté M.H. Trabulsi L.R. Elias W.P. Irino K. Pestana de CastroA.F., 2009. Clonal relationship among atypical enteropathogenic Escherichia coli strains isolated from different animal species and humans. Appl. Environ. Microb. 75:7399–7408.
- PenteadoA.S. Ugrinovich L.A. Blanco J. Blanco M. Blanco J.E. Mora A. Andrade J.R.C. Correa S.S. Pestana de CastroA.F., 2002. Serobiotypes and virulence genes of Escherichia coli strains isolated from diarrheic and healthy rabbits in Brazil. Vet. Microbiol. 89:41–51.
- PlatellJ.L. Johnson J.R. Cobbold R.N. Trott D.J., 2011. Multidrug-resistant extraintestinal pathogenic Escherichia coli of sequence type ST131 in animals and foods. Vet. Microbiol. 153:99–108.
- PoetaP. Radhouani H. Goncalves A. Figueiredo N. Carvalho C. Rodrigues J. Igrejas G., 2010. Genetic characterization of antibiotic resistance in enteropathogenic Escherichia coli carrying extended-spectrum beta-lactamases recovered from diarrhoeic rabbits. Zoonoses Public Hlth 57:162–170.
- ScaifeH.R. Cowan D. Finney J. Kinghorn-Perry S.F. Crook B., 2006. Wild rabbits (Oryctolagus cuniculus) as potential carriers of verocytotoxin-producing Escherichia coli. Vet. Rec. 159:175–178.
- SilvaN. Igrejas G. Figueiredo N. Gonçalves A. Radhouani H. Rodrigues J. Poeta P., 2010. Molecular characterization of antimicrobial resistance in enterococci and Escherichia coli isolates from European wild rabbit (Oryctolagus cuniculus). Sci. Total Environ. 408:4871–4876.
- StamatakisA., 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690.
- SteinslandH. Lacher D.W. Sommerfelt H. Whittam T.S., 2010. Ancestral lineages of human enterotoxigenic Escherichia coli. J. Clin. Microbiol. 48:2916–2924.
- TennantS.M. Tauschek M. Azzopardi K. Bigham A. Bennett-Wood V. Hartland E.L. Qi W. Whittam T.S. Robins-Browne R.M. 2009. Characterisation of atypical enteropathogenic E. coli strains of clinical origin. BMC Microbiol. 9:117–127.