
Abstract
Aim: The presence of di-/multi-meric forms of soluble target in biological samples can interfere in anti-drug antibody (ADA) assays, leading to increased background values and potentially false positivity. The authors investigated the use of the high ionic strength dissociation assay (HISDA) to reduce target interference in two different ADA assays. Results: Interference caused by homodimeric FAP was successfully eliminated to enable cut point determination after applying HISDA. Biochemical experiments confirmed the dissociation of homodimeric FAP after treatment with high ionic strength conditions. Conclusion: HISDA is a promising approach to simultaneously achieve high drug tolerance and reduced interference by noncovalently bound dimeric target molecules in ADA assays without extensive optimization, which is particularly advantageous in routine use.
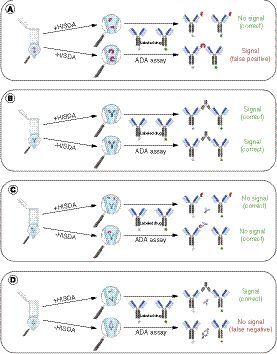
(A) HISDA-induced dissociation of dimeric target into monomers that cannot bridge with ADA assay reagents. The undissociated target dimers in the untreated sample (-HISDA) can lead to false-positive signals by bridging with ADA assay reagents. (B) The application of HISDA has no impact on ADA detectability compared with untreated. (C) The HISDA-induced dissociation of drug–target complexes has no impact on the assay result, since the released dimeric target is subject to dissociation according to (A) into monomers that are unable to bridge with ADA assay reagents. (D) The HISDA-induced dissociation of drug–ADA complexes enables drug-tolerant ADA detection. The undissociated drug–ADA complex is not detectable and therefore leads to a lower drug tolerance in untreated samples.
ADA: Anti-drug antibody; HISDA: High ionic strength dissociation assay.
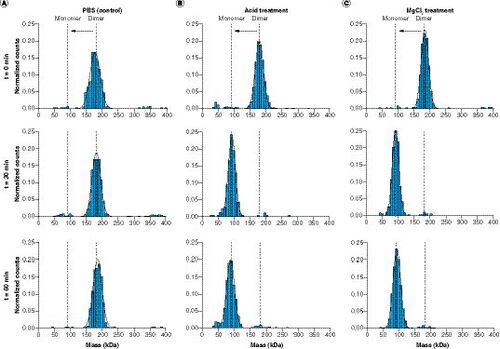
(A) Analysis of undiluted FAP (t = 0 min) and after tenfold dilution and incubation with 1× PBS (phosphate-buffered saline; t = 30 min and t = 60 min). (B) Analysis of FAP before acid treatment (t = 0 min) and after dilution and incubation with 0.1 M glycine-HCl, pH 2.0 (t = 30 min) and after further dilution and incubation using 0.5 M tris(hydroxymethyl)aminomethane-HCl, pH 8.5 (t = 60 min). (C) Analysis of FAP before MgCl2 treatment (t = 0 min) and after tenfold dilution and incubation with MgCl2 solution (t = 30 min) and after further tenfold dilution and incubation using 1× PBS (t = 60 min).
PBS: Phosphate-buffered saline.
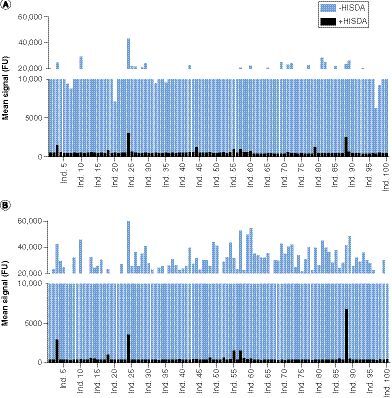
(A) Mean signals in fluorescence units of the analysis of sera from 100 individual donors using the mAb1-anti-drug antibody assay (ADA) with high ionic strength dissociation assay (HISDA; black) and without HISDA (blue). The substrate development and readout conditions (e.g., reader settings, development time and instrument gain) of the two assays were comparable. (B) Mean signals of the analysis of sera from 100 individual donors using the mAb2-ADA assay with HISDA (black) and without HISDA (blue).
FU: Fluorescence units; HISDA: High ionic strength dissociation assay; Ind.: Individual donor.
The analysis of anti-drug antibodies (ADAs) is crucial for the assessment of the safety and efficacy of biopharmaceuticals. However, the presence of dimeric and multimeric soluble targets in biological samples can interfere with the measurement of ADAs, leading to increased background values and potentially resulting in false positivity due to bridging with ADA assay reagents in the case of conventional ADA bridging assays [Citation1–3]. Therefore, the reduction of this interference is essential for unbiased ADA assay results.
Several approaches are available to eliminate the interference when measuring ADAs. Employing masking reagents, such as specific antibodies or recombinant soluble receptors, is one option. The masking reagents bind to the soluble target and abolish the interaction with labeled drug molecules that are used typically as ADA assay reagents [Citation2–5]. The utilization of such masking reagents against the soluble target can provide a solution to the interference issue. However, the development, validation and production of such reagents are a time-consuming and costly process that requires significant resources. Other approaches do exist, such as polyethylene glycol (PEG) precipitation [Citation6] or solid-phase separation of ADA from the soluble target, and do not require the high production cost of tailored reagents. However, these methods pose a risk of under-recovery of ADAs due to potential coprecipitation of ADAs and/or soluble target, as the PEG precipitation is based on the isoelectric point of the complexes as well as on the chosen experimental conditions (e.g., PEG of differing molecular weight, PEG concentration, overall protein content and pH) [Citation4,Citation7,Citation8]. Another way to overcome target interference is the use of the isoelectric target depletion approach [Citation9].
The availability of an alternative, easy-to-use and robust method for elimination of the soluble target interference would be valuable, as it would allow for efficient immunogenicity testing. Even better would be to have the option to overcome not only the soluble target interference challenge but also the fundamental difficulties of achieving highly drug-tolerant ADA assays.
Improvement of drug tolerance is typically achieved by using conditions that disrupt existing ADA–drug complexes to release the ADA and thus enable detection in the ADA bridging assay. Acid dissociation pretreatment is often used for this purpose. Recently, it was reported that the high ionic strength dissociation assay (HISDA) is also suitable for achieving improved drug tolerance [Citation10]. Since both approaches involve a step that disrupts protein–protein interactions, the authors of the current study tested both possibilities for their additional ability to simultaneously reduce the interference caused by a noncovalently bonded homodimeric target.
Here the authors report the results of their investigation regarding the potential use of the typically applied HISDA conditions [Citation10] and acid dissociation conditions based on glycine hydrochloride [Citation10–12] for overcoming dimeric target interference in ADA assays. First, biochemical experiments to test the ability of the two disruptive protocols to dissociate a noncovalently bound homodimeric soluble target were performed to assess the principle suitability of both methods. Subsequently, the authors continued with the HISDA approach and applied it to two different ADA bridging assay examples that showed a pronounced target interference to demonstrate principle proof of applicability.
Experimental section
Positive controls (PCs)
An equimolar mixture of two monoclonal antibodies derived from a murine source directed against the therapeutic monoclonal antibody mAb1 (mAb<mAb1>M; Roche Diagnostics GmbH, Mannheim, Germany) was used in the mAb1-ADA assay (PC1). It was dissolved at 1.33 mg/ml in an aqueous solution of 50 mM potassium phosphate (Merck Chemicals GmbH, Darmstadt, Germany), 150 mM potassium chloride (Merck Chemicals GmbH) and 6.5% sucrose (Merck Chemicals GmbH), pH 7.5.
A monoclonal antibody derived from a murine source directed against the therapeutic monoclonal antibody mAb2 (mAb<mAb2>M; Roche Diagnostics GmbH) was used in the mAb2-ADA assay (PC2). It was dissolved at 3.98 mg/ml in an aqueous solution of 50 mM potassium phosphate, 150 mM potassium chloride and 6.5% sucrose, pH 7.5.
Recombinant soluble dimer
A soluble homodimeric form of FAP (Roche Diagnostics GmbH) consisting of two noncovalently bound subunits, each with a predicted molecular weight of approximately 89 kDa, was used for dissociation experiments. It was dissolved at 3.3 mg/ml in an aqueous solution of 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (Merck Chemicals GmbH), 150 mM sodium chloride (Merck Chemicals GmbH) and 6.5% sucrose, pH 7.5.
Human matrices
Human serum from 100 individual donors was purchased from TRINA Bioreactives AG (Nänikon, Switzerland). 85 individual sera were mixed to establish a human pooled matrix and used as a negative control. 15 out of 100 individual sera were excluded from pooling due to limited sample volume.
mAb1-ADA screening assay (with HISDA)
The following method was previously published [Citation10]. A bridging ELISA was established for the qualitative detection of ADAs directed against the therapeutic monoclonal antibody mAb1. The antibody mAb1 is directed against FAP, which is a serine protease that also occurs in a soluble dimeric form [Citation13]. All incubation steps were performed with shaking at 450 r.p.m. at room temperature. Human pooled serum was used spiked with different concentrations of PC1 (quality controls) and unspiked (negative control). All quality controls, negative controls and samples were analyzed in duplicate measurements. Each run contained one aliquot of high-concentration PC1 (HPC) quality control and low-concentration PC1 (screening LPC [sLPC]) quality control, as well as four aliquots of negative control. Quality controls, negative controls and samples were diluted 1:10 using a 4 M MgCl2*6H2O solution (MgCl2; Merck Chemicals GmbH) and incubated for 30 min (dissociation step). Subsequently, the samples were further diluted 1:10 using LowCross-Buffer (Candor Bioscience GmbH, Wangen, Germany) containing 1.11 μg/ml mAb1-Biotin (Roche Diagnostics GmbH) and 1.11 μg/ml mAb1-digoxigenin (Roche Diagnostics GmbH, Mannheim, Germany). The resulting solution was incubated for another 30 min. The formed immune complexes were then transferred to a streptavidin-coated microtiter plate (MTP; Microcoat Biotechnologie GmbH, Bernried, Germany) at 100 μl/well and incubated for 1 h, followed by three consecutive washing steps using 300 μl 1× phosphate-buffered saline (PBS; Roche Diagnostics GmbH) containing 0.05% Tween20 (Roche Diagnostics GmbH). After washing, 100 μl/well LowCross-Buffer containing 50 mU/ml antidigoxigenin Fab fragments labeled with horseradish peroxidase (Roche Diagnostics GmbH) was added to the MTP and incubated for 1 h. After another three washing steps, 100 μl/well 0.1 M tris(hydroxymethyl)aminomethane (TRIS; Merck Chemicals GmbH), pH 8.5 containing 20 mM 3-(4-hydroxyphenyl)propionic acid (HPPA; Thermo Fisher Scientific, Inc., MA, USA) [Citation14] and 0.02% hydrogen peroxide solution 30% (Merck Chemicals GmbH) were added to the MTP and incubated for 10 min. Fluorescence intensity measurement was performed at an excitation wavelength of 320 nm and an emission wavelength of 400 nm using a BioTek Synergy HTX reader (Agilent, CA, USA).
mAb1-ADA screening assay (without HISDA)
The same procedure was followed as for the mAb1-ADA screening assay (with HISDA) but using LowCross-Buffer instead of 4 M MgCl2*6H2O solution at the dissociation step.
mAb2-ADA screening assay (with HISDA)
The same procedure was followed as for the mAb1-ADA screening assay (with HISDA), except using PC2 instead of PC1 for the preparation of quality controls and mAb2-biotin/digoxigenin (Roche Diagnostics GmbH) instead of mAb1-biotin/digoxigenin. mAb2 is a therapeutic monoclonal antibody that is analogous to mAb1 and binds to FAP but on a different epitope.
mAb1/2-ADA confirmatory assays
Blank matrix samples of 100 individual human donors were analyzed three independent times in a total of 15 separate runs unspiked and spiked with excess drug mAb1 or mAb2 using the respective ADA screening assay (with HISDA). The excess drug concentration was 3 μg/ml in both cases and was selected based on preliminary experiments in which the signal of an HPC was inhibited by an arbitrary value of 40%. The excess drug was added to the respective capture/detection solution containing mAb1/2-biotin and -digoxigenin. Each MTP contained the corresponding unspiked and spiked sample to ensure comparability of the values.
Determination of screening cut point & confirmatory cut point
For the determination of the screening cut point normalization factor, blank matrix samples of 100 individual human donors were analyzed in duplicate in three separate runs using the respective mAb1- or mAb2-ADA screening assay (with HISDA). In a first step, technical outliers were excluded from further calculations if the variation coefficient between the duplicate measurement exceeded 25%. Subsequently, all signals from this analysis were normalized to the run-specific negative control. All of the following statistical tests were performed using the software MedCalc, version 12.7.8 (MedCalc Software bvba, Ostend, Belgium). Outliers were excluded based on values higher than three interquartile ranges by box plot. The Shapiro–Wilk test was used to evaluate the data for normal distribution. Afterward, the non-normally distributed dataset was logarithmically transformed. The screening cut point normalization factor was then calculated with a 5% false-positive rate based on the mean and standard deviation of the logarithmically transformed data and degree of freedom based on the sample size. The plate-specific screening cut point was calculated for every run by multiplying the established normalization factor by the mean signal of negative controls of the plate and was used to classify samples into potentially ADA positive and negative. As a second tier, ADA positive samples from the screening assay were tested in the respective confirmatory assay. If the signal inhibition after the addition of excess drug was greater than the confirmatory cut point, the sample was rated confirmed ADA positive.
For determination of the confirmatory cut point, the signal ratios from the analysis of 100 individual human donors with and without excess drug were calculated as a first step. Subsequently, signal ratios were defined as outliers and excluded from further calculation using a box plot if they fell above three-times the interquartile range above the third quartile or below the first quartile. Afterward, the Shapiro–Wilk test was performed to evaluate the data for normal distribution. The confirmatory cut point was subsequently calculated with a 1% false-positive rate based on the mean and standard deviation of the signal ratios and taking into account the degree of freedom.
Assay sensitivity & quality control samples
Assay sensitivity of the respective ADA screening assay was defined as the serum concentration of PC resulting in a signal at screening cut point. Human pooled serum was spiked with 800 ng/ml PC1 (for mAb1-ADA assay) or 2400 ng/ml PC2 (for mAb2-ADA assay) and serially diluted seven-times in human pooled serum using 1:3 dilution steps. Nine independently prepared calibration curves were analyzed in three runs using the respective mAb1- or mAb2-ADA screening assay (with HISDA). For every curve, sensitivity was calculated by interpolating PC concentration at screening cut point. The mean of all calculated sensitivity values was reported as assay sensitivity. The sLPC was established at an arbitrary concentration providing consistent positive reading above the screening cut point. The HPC was selected at a concentration providing an arbitrary instrument gain value of 40 and was used to enable signal comparison between runs.
Assay sensitivity of the respective ADA confirmatory assay was defined as the lowest PC concentration that confirmed as positive. Confirmed positivity was demonstrated by signal reduction above the value of the confirmatory cut point. The signal reduction was determined using signal ratios from the analysis of the respective PC concentration with and without excess drug. The same experimental procedure was followed as for the determination of assay sensitivity of the screening assays, but using the additional analysis with excess drug. The confirmatory LPC (cLPC) was calculated based on the mean and standard deviation of the mean sensitivity value and degree of freedom.
Mass photometry
Mass photometry experiments were performed using the OneMP mass photometer (Refeyn, Oxford, UK). All experiments were performed at room temperature using ready-to-use sample carrier slides (Refeyn, Oxford, UK) and all dilutions were prepared using 1× PBS unless stated otherwise. A stock solution of FAP was prepared at 1 mg/ml and used in all further steps involving either MgCl2 treatment or acid treatment. For the MgCl2 treatment, the stock solution was diluted 1:10 using 4 M MgCl2*6H2O (dissociation step) or 1× PBS (control step) and incubated for 30 min. Aliquots of both preparations were further diluted 1:10 using the same procedure. The resulting samples were analyzed at all three time points (t = 0 min and 30/60 min after the initial dilution) at a final calculated monomeric FAP concentration in the droplet of 2.5 nM (samples treated with 4 M MgCl2*6H2O) and 5 nM (samples treated with 1× PBS). A mass calibration curve was generated by analysis of three different proteins with known molecular masses of approximately 66 kDa, 145.5 kDa and 194 kDa, respectively (Roche Diagnostics GmbH). Data were collected from the 2.9 μm × 10.8 μm instrument field of view for 100 s (calibration points) and 60 s (samples) at a 1 kHz frame rate using AcquireMP 2.5.0 software (Refeyn, Oxford, UK). Results were reported as normalized counts and calculated by dividing events in each bin by the total number of events. Images were processed using DiscoverMP 2.5.0 software and the following parameters: number of averaged frames: 5, threshold 1: 1.5 and threshold 2: 0.25. For the acid treatment, the same procedure was followed as for the MgCl2 treatment, but with the following changes: the stock solution was diluted 1:40 using 1× PBS and 0.1 M Glycine-HCl pH 2.0 (Merck Chemicals GmbH), incubated for 30 min and measured (dissociation step). An aliquot of the preparation was further diluted 1:2.5 using 1× PBS and 0.5 M TRIS-HCl pH 8.5 (Merck Chemicals GmbH), incubated for 30 min and measured (neutralization step).
Results & discussion
During the development of an ADA bridging assay for the detection of antibodies directed against mAb1, an interference by dimeric soluble target was anticipated. This expected interference caused by soluble homodimeric FAP was confirmed by preliminary experiments and was noticeable in the form of high signal intensities when measuring human sera from healthy individual donors. This outcome made it impossible to continue developing the assay because of the difficulty in distinguishing between false-positive signals caused by homodimeric FAP and ADA-derived signals ().
To overcome such soluble target interference, different mitigation strategies are available, such as the usage of anti-target antibodies that compete with the therapeutic drug for binding, solid-phase separation, PEG precipitation and isoelectric target depletion [Citation2–5,Citation9]. Due to known disadvantages of some of these methods such as limited effectiveness, impact on ADA detectability, assay runtime and low throughput, the authors were particularly interested in the implementation of a disruptive step to the assay as an attempt to eliminate the target interference while maintaining the ADA-drug binding properties.
The use of disrupting conditions such as those used in HISDA [Citation10] has the potential to reduce the noncovalent interactions within the dimeric soluble target, resulting in a dissociation of the soluble target into monomers and thus reducing interference (). In addition, it was previously shown that the HISDA protocol enables the detection of ADAs in the presence of high concentrations of drug molecules [Citation10]. Therefore, the use of one assay protocol based on HISDA has the potential to solve two coexisting challenges at the same time by enabling drug-tolerant immunogenicity testing and potentially overcoming interference by dimeric soluble target molecules.
To test the hypothesis that a disruptive step would be applicable in the described case of soluble homodimeric FAP, experiments using mass photometry were performed as a first step. The purpose of these biochemical experiments was to verify the conversion of the homodimeric form of FAP to a monomeric form. The monomeric FAP was expected to be unable to bridge with ADA assay reagents and subsequently could not generate false-positive results in the ADA bridging assay.
Two disruptive principles were used to simulate assay conditions in mass photometry experiments: treatment with MgCl2 solution to represent the previously described HISDA [Citation10] and a commonly used generic acid-based protocol. As a surrogate drug target, recombinant homodimeric FAP was used in the following experiments. In a first assessment, the analysis of untreated FAP resulted in a main peak at a molecular mass of approximately 180 kDa (). This result was consistent with two-times the calculated monomeric molecular mass of the recombinant FAP of approximately 89 kDa. The ratio of the main peak signal to the overall signal indicated a dimer content of >90% in the untreated sample and confirmed the high purity of the material with almost no monomer content. After sample treatment with MgCl2 solution and incubation for 30 min, analogous to the HISDA assay protocol, a shift of the main peak to approximately 90 kDa was observed (). This result was again in line with the calculated molecular mass of the monomeric form of FAP and, importantly, did not change after further 1:10 dilution using 1× PBS and incubation for another 30 min, indicating that no reassociation of the monomers occurred. In addition, FAP that was treated with 1× PBS instead of MgCl2 solution showed no shift of the main dimer peak (). Therefore, the disappearance of the dimer peak and the appearance of the monomer peak at the expected molecular mass in the MgCl2-treated sample confirmed that the FAP dimer dissociated into its monomers due to the high ionic strength conditions. This observation was consistent with the expectation that high ionic strength conditions can influence the stability and disruption of dimers and multimers [Citation15–18].
A similar experiment was performed using low-pH conditions. After performing the dissociation step using acidic conditions, the initial result was comparable to that of the MgCl2 treatment run and appeared in the form of FAP monomerization, which was expected (). Moreover, after the neutralization step, no re-formation of dimers or aggregation was observed. The results therefore suggested that both methods led to a similar result and indicated the principle suitability of both methods.
In both cases, it is nevertheless theoretically possible that FAP monomers may re-form to dimers over a longer period of time. However, since the assay protocols use relatively short incubation times of each 30 min each at the two relevant steps, any potential dimer re-formation after that time period is not expected to have any significant impact on the assay performance.
It has been previously demonstrated that the use of acidic pH could reduce the issue of target interference in ADA assays [Citation2]. On the other hand, an opposing report indicated that acid dissociation steps could lead to increased target dimerization and consequently false-positive results [Citation6]. To minimize such an undesired effect, it is necessary to optimize the parameters of the acid treatment step, especially the pH and incubation time. However, the optimized parameters may not reflect the ideal condition for the dissociation of ADA–drug complexes, which in turn could lead to reduced drug tolerance. Therefore, it could be challenging to optimize an acid dissociation step simultaneously for the dual purpose of reducing target interference and achieving high drug tolerance. This makes the acid-based approach less universally applicable compared with HISDA, which could be used to achieve high drug tolerance [Citation10] and could minimize target interference without the need for extensive optimization. The authors therefore decided to focus on testing the HISDA approach further, instead of the acid treatment, and continued with applying it to two different ADA assay examples.
The measurement of 100 individual human sera using the mAb1-ADA screening assay (with HISDA) resulted in mean signals ranging from 481 to 3085 fluorescence units (FU; average: 653 FU), compared with mean signals ranging from 6275 to 43,179 FU (average: 16,359 FU) in the initial mAb1-ADA screening assay without HISDA (). This promising signal reduction confirmed the previous results of the mass photometry experiments by indicating reduced interference due to the conversion of homodimeric FAP into its monomers, which could no longer bridge with ADA assay reagents. Based on these screening runs using individual sera after applying HISDA, the screening cut point normalization factor was calculated as 1.13. In this context, the determination of the confirmatory cut point was also fulfilled and resulted in a value of 31.6%. Subsequently, experiments for the determination of assay sensitivity were performed for the ADA screening assay and corresponding confirmatory assay. The resulting values were 0.2 ng/ml and 0.6 ng/ml for the screening and confirmatory assays, respectively. Then, the concentrations for sLPC and HPC were set to 0.4 ng/ml and 6.6 ng/ml, respectively. Conclusively, the cLPC concentration was calculated as 1.2 ng/ml. Ensuing assay reproducibility was then assessed by calculating the precision of interassay runs using data from HPC analysis and the corresponding inhibited signal values of the HPC. Based on the results from a total of 22 runs, the precision of HPC was 11.4% and the precision of HPC inhibition was 10.4%.
The overall result of this assay establishment indicated that the use of HISDA has the potential to reliably eliminate the target interference to enable ADA-specific cut point determination and that both the screening assay and confirmatory assay run in a reproducible way that complies with the US FDA's recommendations [Citation19].
To test the usage of HISDA further, a second ADA bridging assay was established. The reagents used in this assay were based on a different therapeutic monoclonal antibody (mAb2), that also binds to FAP but on a different epitope. In this second ADA assay example, the assay variant without HISDA was initially tested. The measurement was performed using the same panel of individual human sera analogous to the mAb1-ADA assay and resulted in mean signals ranging from 10,619 to 63,393 FU (average: 29,571 FU), confirming significant target interference (). After applying HISDA, mean signals decreased and ranged from 308 to 6,883 FU (average: 541 FU; ). The subsequent determination of the screening cut point normalization factor and the confirmatory cut point resulted in values of 1.23 and 22%, respectively. Corresponding assay sensitivities were both equally calculated as 2.1 ng/ml. The concentrations for sLPC, HPC and cLPC were 3.4 ng/ml, 24 ng/ml and 3.4 ng/ml, respectively. Assay reproducibility assessment resulted in an HPC precision of 14.9% and a corresponding precision of HPC inhibition of 2.7%. The results of the second established ADA assay also indicated reduced target interference by applying HISDA, which makes the assay applicable, and were in line with the findings from the biochemical experiments and with the data generated from the mAb1-ADA assay.
The advantages and disadvantages of available mitigation methods for reducing dimeric and multimeric target interference in ADA bridging assays have been extensively discussed [Citation4]. The HISDA approach shown here solves several disadvantages of alternative methods such as the time-consuming generation of masking reagents, low sample throughput and unknown impact on ADA detectability. In return, the method offers an easy-to-use protocol that enables high drug-tolerant immunogenicity testing and reduction of dimeric target interference.
Conclusion
The use of HISDA for the reduction of soluble target interference in ADA bridging assays has been demonstrated to be a useful tool in cases where dimeric or multi-meric targets are held together by noncovalent interactions that are sensitive to high salt concentrations. Due to the simple feasibility of the method, it represents an attractive alternative to the conventional approaches to mitigating dimeric target interference and is particularly advantageous in routine use. The monomers that result from the dimer dissociation do not bridge with ADA assay reagents and therefore do not generate false-positive assay results. It is important to note that the HISDA approach, as well as acid dissociation and in general all protocols that dissociate protein–protein complexes, can be applied only to noncovalent dimeric and potentially multi-meric targets. However, as the majority of dimeric/multimeric endogenous proteins that are used as drug targets belong to this group, this approach has the potential for broad applicability in the bioanalytical field.
The HISDA approach offers several advantages over alternative methods in applicable cases. First, the protocol is relatively easy to implement without the need for extensive optimization. Second, HISDA has been shown to greatly improve drug tolerance [Citation10]. In addition, it is considered to be a gentle disruptive method that maintains ADA detectability [Citation10]. Overall, the use of the HISDA approach in this study demonstrated a reduction of soluble target interference in two different ADA assay examples and therefore offers a viable alternative over more complex methods that may achieve a similar result. However, since the effectiveness of target interference reduction is dependent on the structure of the soluble target, the applicability should be assessed on a case-by-case basis.
Immunogenicity testing will remain an essential component of the development of biotherapeutic products. The need for ADA-specific methods that are not affected by dimeric or multimeric target interference is therefore crucial. The increasing use of drug combinations and switch of patients from one therapeutic drug to another, both of which bind the same target, bears the potential to lead to false-positive results, even in cases of monomeric targets, due to artificial dimerization by bivalent therapeutic drugs [Citation9,Citation20]. As a result, there is a growing demand for methods that are universally applicable and easy to implement in a routine setting. The use of HISDA proved suitable for dissociating ADA–drug complexes as well as for dissociating a noncovalently bound dimeric target, without requiring time and labor-intensive adaptations or optimization. Further evaluations and examples might show the potential generic applicability for overcoming interference caused by noncovalent di- and multi-meric targets. The easy-to-use two-step protocol makes it particularly advantageous for routine testing and has the potential to be easily automated.
Summary points
The existence of dimeric or multimeric forms of soluble drug targets in biological samples can interfere in anti-drug antibody (ADA) assays by bridging the ADA assay reagents, leading to increased background values and potentially resulting in false positivity.
The reduction of soluble target interference is essential in order to obtain unbiased ADA assay results, which are required for assessment of the safety and efficacy of biopharmaceuticals.
One hundred individual human sera were screened with two different one-step ADA bridging assays, each with and without the application of the high ionic strength dissociation assay (HISDA) approach, to investigate the potential reduction of interference caused by soluble noncovalently bound homodimeric FAP.
Biochemical experiments were performed to test for the dissociation of a homodimeric form of FAP into its monomers after treatment with high ionic strength conditions.
The use of HISDA eliminated soluble homodimeric FAP interference and enabled ADA-specific cut point determination in two different ADA bridging assays.
Biochemical experiments confirmed the conversion of a homodimeric form of FAP to a monomeric form after treatment with high ionic strength conditions.
The use of HISDA represents a promising approach to simultaneously achieving high drug tolerance and reducing interference caused by noncovalently bound dimeric target molecules in ADA bridging assays without the need for extensive method optimization.
The method offers advantages such as simple implementation and the preservation of ADA detectability and is particularly advantageous in routine use.
Acknowledgments
was created with BioRender.com. The authors acknowledge Hubert Kettenberger (Roche Diagnostics GmbH), Thomas Emrich (Roche Diagnostics GmbH), Steven Challand (F. Hoffmann-La Roche AG), Kerstin Völse (Microcoat Biotechnologie GmbH), Christopher Georgiev (Microcoat Biotechnologie GmbH) and Orsolya Gergely (Microcoat Biotechnologie GmbH).
Financial & competing interests disclosure
All authors are employees of Roche Diagnostics GmbH. Some authors are owners of Roche bonus shares and profit certificates. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- Dai S , SchantzA, Clements-EganA, CannonM, ShankarG. Development of a method that eliminates false-positive results due to nerve growth factor interference in the assessment of fulranumab immunogenicity. AAPS J.16(3), 464–477 (2014).
- Liao K , MeyerE, LeeTN, LoercherA, SikkemaD. Inhibition of interleukin-5 induced false positive anti-drug antibody responses against mepolizumab through the use of a competitive blocking antibody. J. Immunol. Methods441, 15–23 (2017).
- Chen J , KendraK, TorriA, SumnerG. Overcoming multimeric target interference in a bridging immunogenicity assay with soluble target receptor, target immunodepletion and mild acidic assay pH. Bioanalysis12(15), 1071–1085 (2020).
- Zhong ZD , Clements-EganA, GorovitsBet al. Drug target interference in immunogenicity assays: recommendations and mitigation strategies. AAPS J.19(6), 1564–1575 (2017).
- Zhong ZD , DinnogenS, HokomMet al. Identification and inhibition of drug target interference in immunogenicity assays. J. Immunol. Methods355(1-2), 21–28 (2010).
- Zoghbi J , XuY, GrabertR, TheobaldV, RichardsS. A breakthrough novel method to resolve the drug and target interference problem in immunogenicity assays. J. Immunol. Methods426, 62–69 (2015).
- Ingham KC . Precipitation of proteins with polyethylene glycol. Methods Enzymol.182, 301–306 (1990).
- Polson A , PotgieterGM, LargierJF, MearsGE, JoubertFJ. The fractionation of protein mixtures by linear polymers of high molecular weight. Biochim. Biophys. Acta82, 463–475 (1964).
- Jordan G , Moheysen-ZadehM, DahlU, StaackRF. Novel isoelectric target depletion (ITaD) protocol reduces the need for specific reagents for immunogenicity testing. Bioanalysis14(10), 725–735 (2022).
- Jordan G , PohlerA, GuilhotF, ZaspelM, StaackRF. High ionic strength dissociation assay (HISDA) for high drug tolerant immunogenicity testing. Bioanalysis12(12), 857–866 (2020).
- Hoffmann E , JordanG, LauerMet al. Generation, characterization, and quantitative bioanalysis of drug/anti-drug antibody immune complexes to facilitate dedicated in vivo studies. Pharm. Res.36(9), 129 (2019).
- Myler HA , McVayS, KratzschJ. Troubleshooting PEG-hGH detection supporting pharmacokinetic evaluation in growth hormone deficient patients. J. Pharmacol. Toxicol. Methods61(2), 92–97 (2010).
- O'Brien P , O'ConnorBF. Seprase: an overview of an important matrix serine protease. Biochim. Biophys. Acta1784(9), 1130–1145 (2008).
- Jordan G , StubenrauchKG, HeinrichJ, StaackRF. 3-(4-Hydroxyphenyl)propionic acid: the forgotten detection substrate for ligand-binding assay-based bioanalysis. Bioanalysis9(4), 407–418 (2017).
- Couthon F , ClottesE, VialC. High salt concentrations induce dissociation of dimeric rabbit muscle creatine kinase. Physico-chemical characterization of the monomeric species. Biochim. Biophys. Acta1339(2), 277–288 (1997).
- Jilaveanu LB , ZitoCR, OliverD. Dimeric SecA is essential for protein translocation. Proc. Natl Acad. Sci. USA102(21), 7511–7516 (2005).
- Lee HS , KimJS, ShimKet al. Dissociation/association properties of a dodecameric cyclomaltodextrinase. Effects of pH and salt concentration on the oligomeric state. FEBS J.273(1), 109–121 (2006).
- Gronau G , QinZ, BuehlerMJ. Effect of sodium chloride on the structure and stability of spider silk's N-terminal protein domain. Biomater. Sci.1(3), 276–284 (2013).
- US FDA . Immunogenicity Testing of Therapeutic Protein Products – Developing and Validating Assays for Anti-drug Antibody Detection.MD, USA (2019).
- Nishidate M , ShibaharaN, HayasakaAet al. Validation of a method to analyze size distribution of crovalimab-complement C5-eculizumab complexes in human serum. Bioanalysis14(13), 935–947 (2022).