
Abstract
Background: Increasing rates of acquired resistance have justified the critical need for novel antimicrobial drugs. One viable concept is the modification of known drugs. Methods & results: 21 mafenide-based compounds were prepared via condensation reactions and screened for antimicrobial efficacy, which demonstrated promising activity against both Gram-positive and Gram-negative pathogens, pathogenic fungi and mycobacterial strains (minimum inhibitory concentrations from 3.91 μM). Importantly, they retained activity against a panel of superbugs (methicillin- and vancomycin-resistant staphylococci, enterococci, multidrug-resistant Mycobacterium tuberculosis) without any cross-resistance. Unlike mafenide, most of its imines were bactericidal. Toxicity to HepG2 cells was also investigated. Conclusion: Schiff bases were significantly more active than the parent drug, with iodinated salicylidene and 5-nitrofuran/thiophene-methylidene scaffolds being preferred in identifying the most promising drug candidates.
Graphical abstract

There is an urgent need worldwide to combat microbial drug resistance. We can distinguish various approaches for the production of new anti-infective drugs, including the reuse of known and later abandoned drugs and the modification of effective scaffolds.
Sulfonamides, also called sulfa drugs, are synthetic antibiotics used for the treatment of bacterial infections and some fungal and protozoal infections. They were the first broadly effective antibacterials, which paved the way for the antibiotic revolution in medicine [Citation1]. They directly interfere with the synthesis of dihydropteroic acid, a precursor of tetrahydrofolic acid, the coenzymatic form of folic acid that bacteria need for folate synthesis and ultimately purine and nucleic acids [Citation2]. Because they inhibit the growth and multiplication of bacteria, they have a bacteriostatic effect. Sulfanilamides became the basis for the development of other types of medicinally important compounds with a varied spectrum of biological activities.
Mafenide (MAF), also called homosulfanilamide (), was synthesized in the USA in 1938 [Citation3] for the treatment of post-burn infections [Citation4]. Its molecule differs from classical sulfonamides, because it has an aminomethyl group in the para position on the benzene ring instead of the simple amino group of sulfonamides (). The first extensive use of mafenide was in the treatment of burn wound infections [Citation5] and it is still utilized as a topical remedy [Citation6]. Mafenide acetate (MFA; the most frequently used MAF salt, Sulfamylon®) exhibits antibacterial/bacteriostatic action against many Gram-positive and Gram-negative microorganisms, including Staphylococcus aureus and Pseudomonas aeruginosa, and it is used to treat infections caused especially by Gram-negative microorganisms [Citation7,Citation8], although with comparatively higher numerical values of minimum inhibitory concentration (MIC). In addition, it has mild anti-inflammatory [Citation9] and antifungal [Citation10] capacities.
PABA: 4-aminobenzoic acid.
Its mechanism of action was expected to be the same as that of sulfonamides, i.e., interfering with 4-aminobenzoic acid (PABA; Figure 1) utilization and thus inhibiting dihydropteroate synthase. However, MAF did not inhibit the synthesis of dihydropteroate by dihydropteroate synthase in lysates of either P. aeruginosa or Escherichia coli [Citation11]. Generally, the mechanism of action is distinct from ‘classical' sulfonamides, as sulfonamide-resistant bacteria remain susceptible to MAF [Citation12]. MAF has been demonstrated as an inhibitor of dihydrofolate synthase [Citation9]. It might also indirectly inactivate the folic acid transport system [Citation13]. The disruption of nucleotide biosynthesis was also reported [Citation7]. The mechanism of mafenide cytotoxicity involves selective inhibition of the de novo bases synthesis pathway [Citation14]. MAF also inhibits a broad spectrum of carbonic anhydrases from various organisms [Citation12,Citation15]. Most importantly, from a clinical point of view, MAF is not antagonized by the presence of PABA, serum, pus or exudates, and the activity is not altered by the pH of the environment. This drug does not induce mutations [Citation12].
The high antimicrobial activity of copper nanoparticles against a broad range of pathogenic microbes and their relatively low toxicity to humans led to the development of MFA loaded in copper nanocomposite (CuNP). MIC for MFA–CuNPs was four-times better (MIC 12.5 μg/ml for MFA with CuNPs, 50 μg/ml for MFA only) [Citation16]. MFA was also evaluated for its in vitro activity against bacterial and fungal biofilms but showed only a minimal activity against the biofilms tested [Citation17].
Carbamate derivatives of the aminomethyl group of mafenide were synthesized as potential antimicrobial agents. However, none of the new compounds was active in antimicrobial or antiviral screening [Citation18].
Owing to the imine bond (R1R2C=NR3) of Schiff bases, these have a wide spectrum of pharmacological properties and are an important class of organic compounds in medicinal chemistry. Various studies have reported sulfonamide-derived Schiff bases as urease inhibitors [Citation1], antibacterial [Citation19,Citation20] or antitubercular [Citation21] compounds. MAF-derived imines and their coumarin hybrids have been reported as carbonic anhydrase inhibitors and cytotoxic agents [Citation22].
Based on the previous experience with imine derivatives of PABA [Citation23] and ‘classical' antibacterial sulfonamides [Citation24,Citation25], we also designed and prepared Schiff bases from MAF as potential antibacterial, antimycobacterial, antifungal and cytotoxic agents.
Experimental
Chemistry
General
All the reagents and solvents were purchased from Merck (Darmstadt, Germany), Penta Chemicals (Prague, Czech Republic), Avantor (Stříbrná Skalice, Czech Republic), and Lach-Ner (Neratovice, Czech Republic). They were used as received without any purification. The reactions and the purity of the products were monitored by thin layer chromatography (TLC) using a mixture with a ratio of dichloromethane to methanol (MeOH) and n-hexane of 4:1:1 (v/v/v) as the eluent. Plates were coated with 0.2 mm Merck 60 F254 silica gel (Merck Millipore, Darmstadt, Germany) and were visualized by UV irradiation (254 nm). The melting points were determined on a Melting Point B-560 apparatus (BÜCHI, Flawil, Switzerland) using open capillaries. The reported values are uncorrected. Infrared spectra (IR) were recorded on a FT–IR spectrometer using attenuated total reflectance (ATR)-Ge method (Nicolet 6700 FT–IR, Thermo Fisher Scientific, MA, USA) in the range 600–4000 cm-1. The nuclear magnetic resonance (NMR) spectra were measured in dimethyl sulfoxide (DMSO)-d6, or acetone-d6 at ambient temperature using a Varian V NMR S500 instrument (500 MHz for 1H and 126 MHz for 13C; Varian Corp., CA, USA) or a JNM-ECZ 600R (600 MHz for 1H a 151 MHz for 13C; JEOL, Tokyo, Japan). The chemical shifts δ are given in ppm and were referred indirectly to tetramethylsilane via signals of DMSO-d6 (2.50 for 1H and 39.51 for 13C spectra) or acetone-d6 (1.91 or 1H, 29.8 and 206.3 for 13C spectra). The coupling constants (J) are reported in Hertz. Elemental analysis (C, H, N) was performed using an automatic microanalyzer, Vario MICRO Cube Element Analyzer (Elementar Analysensysteme, Hanau, Germany). The calculated and found values are given as percentages.
LogP values, logarithm of partition coefficient (octan-1-ol/water), were calculated with the ChemOffice 2019 program (version 20.0, CambridgeSoft, MA, USA) using Crippen's fragmentation approach.
The identity of the known compounds was established using NMR (1H and 13C) and IR spectroscopy by the comparison with the previously reported data. The purity was checked by melting points measurement and elemental analysis. The compounds were considered as pure if they agree within ±0.4% with the theoretical values.
Synthesis
Synthesis of precursor
2-Formyl-4,6-diiodophenyl acetate was synthesized as follows. 3,5-Diiodosalicylaldehyde (1 mmol) was dissolved in dry dichloromethane (7 ml) followed by the addition of triethylamine (1.5 mmol). After cooling to 0°C, acetyl chloride (1.2 mol) was added in one portion under vigorous stirring. The mixture was let to stir without external cooling for 1 h (based on TLC indication of a complete reaction; mobile phase: n-hexane: ethyl acetate 4:1, v/v). It was evaporated to dryness under reduced pressure and transferred into a separation funnel using ethyl acetate and water. The organic layer was washed with water, 10% aqueous sodium carbonate, 0.1 M aqueous hydrochloric acid and saturated brine. The organic phase was dried over anhydrous sodium sulfate. The filtrate was concentrated under reduced pressure, and n-hexane was added to start precipitation. After 24 h at +4°C, the suspension was filtered off to yield the precursor.
2-Formyl-4,6-diiodophenyl acetate. Yellowish solid; yield 93%; mp 142.5°C. IR (ATR): 3059, 1754, 1683, 1566, 1548, 1436, 1402, 1379, 1362, 1227, 1192, 1133, 1094, 1010, 907, 880, 733, 701, 669 cm-1. 1H NMR (600 MHz, DMSO-d6): δ 9.82 (1H, s, COH), 8.50 (1H, d, J = 2.1 Hz, H6), 8.16 (1H, d, J = 2.1 Hz, H4), 2.36 (3H, s, CH3). 13C NMR (151 MHz, DMSO): δ 189.59, 168.72, 151.76, 151.40, 140.72, 130.82, 97.52, 93.45, 21.37. Elemental analysis for C9H6I2O3 (415.95); calculated: C, 25.99; H, 1.45, found C, 25.97; H, 1.42.
Imines
Method A
Mafenide acetate (1 mmol; 246.3 mg) was dissolved in 8 ml of MeOH, and then an appropriate aldehyde (1.1 mmol) was added in one portion. The reaction mixture was heated under reflux for 3 h and then stirred to cool down to room temperature overnight. Then, the reaction mixture was stored at -20°C for 2 h. The precipitate, if present, was collected by filtration. When no spontaneous precipitation was observed, the methanolic solution was diluted by water to initiate precipitation and then stored for 1 h at +4°C. The resulting precipitate was collected by filtration, washed by cold water followed by diethyl ether and dried. The products were crystallized from MeOH if necessary.
Method B
Mafenide acetate (1 mmol; 246.3 mg) was dissolved in 8 ml of MeOH, and then an appropriate aldehyde or ketone (1.1 mmol) was added in one portion, followed by nickel(II) nitrate hexahydrate (0.1 mmol, 29.8 mg). The reaction mixture was stirred for 12 h at room temperature and then cooled to -20°C. The precipitate was filtered off, washed thoroughly by cold water and diethyl ether and dried.
Synthesis of the reduced analog 4f
3,5-Diiodosalicylaldehyde (411.3 mg, 1.1 mmol) was dissolved in 15 ml of MeOH, followed by the addition of mafenide acetate (1 mmol; 246.3 mg). Then, sodium cyanoborohydride (100 mg; 1.6 mmol) was added. The mixture was stirred for 48 h at room temperature, and spontaneous precipitation was observed. Then, the reaction mixture was diluted with distilled water and stirred for 30 min at room temperature. The precipitate was filtered off, washed using water and dried. The product was recrystallized from ethyl acetate.
(E)-4-{[(2-Hydroxybenzylidene)amino]methyl}benzenesulfonamide 2a
Yellowish solid; yield 76% (method A); mp 158.4°C (162°C [Citation26]). IR (ATR): 3345, 3254, 1630, 1579, 1498, 1434, 1331, 1277, 1154, 1119, 1094, 1063, 1017, 1006, 903, 837, 810, 777, 764, 731, 710, 655 cm-1. 1H NMR (500 MHz, DMSO-d6): δ 13.26 (1H, s, OH), 8.75 (1H, s, N=CH), 7.84–7.81 (2H, m, H2, H6), 7.54–7.51 (2H, m, H3, H5), 7.50 (1H, dd, J = 7.6, 1.7 Hz, H6′), 7.38–7.33 (3H, m, SO2NH2, H4′), 6.93 (1H, td, J = 7.5, 1.1 Hz, H5′), 6.89 (1H, dd, J = 8.3, 1.1 Hz, H3′), 4.88 (2H, s, CH2). 13C NMR (126 MHz, DMSO): δ 167.39, 160.56, 143.18, 142.88, 132.76, 132.01, 128.36, 126.17, 118.96, 118.89, 116.65, 61.64. Elemental analysis for C14H14N2O3S (290.34); calculated: C, 57.92; H, 4.86; N, 9.65, found C, 58.01; H, 4.83; N, 9.69.
(E)-4-{[(5-Fluoro-2-hydroxybenzylidene)amino]methyl}benzenesulfonamide 2b
Yellow solid; yield 92% (method A); mp 141.1°C. IR (ATR): 3334, 3252, 1640, 1589, 1498, 1480, 1321, 1309, 1269, 1254, 1225, 1167, 1153, 1094, 1053, 874, 846, 811, 782, 744, 682, 664 cm-1. 1H NMR (600 MHz, DMSO-d6): δ 12.90 (1H, s, OH), 8.68 (1H, s, N=CH), 7.81–7.78 (2H, m, H2, H6), 7.51–7.48 (2H, m, H3, H5), 7.35 (1H, dd, J = 9.0, 3.3 Hz, H6′), 7.20–7.16 (1H, m, H4′), 7.31 (2H, s, SO2NH2), 6.88 (1H, d, J = 9.0, 4.5 Hz, H3′), 4.86 (2H, s, CH2). 13C NMR (151 MHz, DMSO): δ 166.40, 157.09, 155.30 (d, J = 234.1 Hz), 143.59, 143.05, 128.77, 126.54, 120.04 (d, J = 23.2 Hz), 119.42 (d, J = 7.3 Hz), 118.33 (d, J = 8.1 Hz), 117.20 (d, J = 23.4 Hz), 62.10. Elemental analysis for C14H13FN2O3S (308.33); calculated: C, 54.54; H, 4.25; N, 9.09, found C, 54.59; H, 4.30; N, 9.16.
(E)-4-{[(5-Chloro-2-hydroxybenzylidene)amino]methyl}benzenesulfonamide 2c
Light yellow solid; yield 85% (method A); mp 162.5°C (161°C [Citation27]). IR (ATR): 3334, 3259, 1635, 1577, 1480, 1412, 1370, 1329, 1310, 1282, 1169, 1156, 1100, 1034, 901, 813, 783, 772, 709, 671, 654 cm-1. 1H NMR (500 MHz, DMSO-d6): δ 13.28 (1H, s, OH), 8.73 (1H, s, N=CH), 7.84–7.81 (2H, m, H2, H6), 7.61 (1H, d, J = 2.7 Hz, H6′), 7.55–7.51 (2H, m, H3, H5), 7.38 (1H, dd, J = 8.8, 2.7 Hz, H4′), 7.34 (2H, s, SO2NH2), 6.92 (1H, d, J = 8.8 Hz, H3′), 4.89 (2H, s, CH2). 13C NMR (126 MHz, DMSO): δ 166.00, 159.41, 143.24, 142.56, 132.37, 130.76, 128.40, 126.17, 122.28, 120.05, 118.73, 61.57. Elemental analysis for C14H13ClN2O3S (324.78); calculated: C, 51.77; H, 4.03; N, 8.63, found C, 51.81; H, 3.99; N, 8.67.
(E)-4-{[(5-Bromo-2-hydroxybenzylidene)amino]methyl}benzenesulfonamide 2d
Light yellow solid; yield 82% (method A); mp 171.5°C (173°C [Citation27]). IR (ATR): 3334, 3259, 1634, 1570, 1477, 1412, 1369, 1328, 1310, 1281, 1185, 1156, 1100, 1033, 899, 822, 812, 785, 772, 695, 670, 646, 626 cm-1. 1H NMR (600 MHz, DMSO-d6): δ 13.27 (1H, s, OH), 8.69 (1H, s, N=CH), 7.80–7.77 (2H, m, H2, H6), 7.69 (1H, d, J = 2.5 Hz, H6′), 7.51–7.48 (2H, m, H3, H5), 7.45 (1H, dd, J = 9.0, 2.5 Hz, H4′), 7.30 (2H, s, SO2NH2), 6.84 (1H, d, J = 8.7 Hz, H3′), 4.85 (2H, s, CH2). 13C NMR (151 MHz, DMSO): δ 166.32, 160.23, 143.63, 142.92, 135.52, 134.07, 128.77, 126.54, 121.05, 119.56, 109.96, 61.91. Elemental analysis for C14H13BrN2O3S (369.23); calculated: 45.54; H, 3.55; N, 7.59, found C, 45.58; H, 3.56; N, 7.62.
(E)-4-{[(2-Hydroxy-5-iodobenzylidene)amino]methyl}benzenesulfonamide 2e
Yellow solid; yield 90% (method A); mp 172°C. IR (ATR): 3331, 3233, 1634, 1599, 1478, 1409, 1347, 1332, 1273, 1157, 1095, 1046, 911, 836, 813, 776, 722, 694, 637, 625 cm-1. 1H NMR (600 MHz, DMSO-d6): δ 13.28 (1H, s, OH), 8.67 (1H, s, N=CH), 7.82 (1H, d, J = 2.4 Hz, H6′), 7.80–7.75 (2H, m, H2, H6), 7.58 (1H, dd, J = 8.7, 2.4 Hz, H4′), 7.50–7.47 (2H, m, H3, H5), 7.30 (2H, s, SO2NH2), 6.71 (1H, d, J = 8.6 Hz, H3′), 4.84 (2H, s, CH2). 13C NMR (151 MHz, DMSO): δ 166.34, 160.80, 143.61, 142.94, 141.16, 140.03, 128.77, 126.54, 121.71, 119.93, 80.55, 61.89. Elemental analysis for C14H13IN2O3S (416.23); calculated: 40.40; H, 3.15; N, 6.73, found C, 40.38; H, 3.14; N, 6.65.
(E)-4-{[(3,5-Difluoro-2-hydroxybenzylidene)amino]methyl}benzenesulfonamide 2f
Yellow solid; yield 89% (method A); mp 218.3°C. IR (ATR): 3337, 3250, 1638, 1602, 1485, 1405, 1329, 1292, 1267, 1165, 1153, 1126, 1091, 1040, 988, 862, 816, 787, 738, 672, 660, 621 cm-1. 1H NMR (600 MHz, DMSO-d6): δ 13.56 (1H, s, OH), 8.72 (1H, s, N=CH), 7.81–7.78 (2H, m, H2, H6), 7.52–7.48 (2H, m, H3, H5), 7.41–7.36 (1H, m, H6′), 7.32 (2H, s, SO2NH2), 7.24–7.21 (1H, m, H4′), 4.89 (2H, s, CH2). 13C NMR (151 MHz, DMSO): δ 166.50, 153.34 (dd, J = 236.1, 10.8 Hz), 150.93 (dd, J = 246.2, 12.1 Hz), 147.50 (d, J = 13.6 Hz), 143.73, 142.53, 128.83, 126.58, 119.86 (dd, J = 8.9, 5.6 Hz), 112.58 (dd, J = 24.9, 3.7 Hz), 108.38 (dd, J = 27.9, 22.0 Hz), 61.15. Elemental analysis for C14H12F2N2O3S (326.32); calculated: C, 51.53; H, 3.71; N, 8.58, found C, 51.48; H, 3.74; N, 8.61.
(E)-4-{[(3,5-Dichloro-2-hydroxybenzylidene)amino]methyl}benzenesulfonamide 2g
Yellow solid; yield 84% (method A); mp 206°C (199°C [Citation27]). IR (ATR): 3337, 3259, 3072, 1634, 1574, 1456, 1140, 1410, 1376, 1327, 1293, 1275, 1255, 1186, 1172, 1152, 1091, 1040, 879, 857, 814, 748, 736, 721, 702, 670 cm-1. 1H NMR (500 MHz, DMSO-d6): δ 14.74 (1H, s, OH), 8.75 (1H, s, N=CH), 7.85–7.82 (2H, m, H2, H6), 7.64 (1H, d, J = 2.7 Hz, H6′), 7.58–7.53 (3H, m, H3, H5, H4′), 7.36 (2H, s, SO2NH2), 4.94 (2H, s, CH2). 13C NMR (126 MHz, DMSO): δ 166.49, 159.92, 143.56, 141.52, 132.65, 130.46, 128.52, 126.27, 123.23, 119.70, 118.58, 58.76. Elemental analysis for C14H12Cl2N2O3S (359.22); calculated: C, 46.81; H, 3.37; N, 7.80, found C, 46.85; H, 3.40; N, 7.76.
(E)-4-{[(3,5-Dibromo-2-hydroxybenzylidene)amino]methyl}benzenesulfonamide 2h
Yellow solid; yield 92% (method A); mp 211°C (198°C [Citation27]). IR (ATR): 3330, 1640, 1594, 1489, 1409, 1322, 1310, 1205, 1161, 1131, 1099, 1020, 915, 865, 839, 810, 730, 681, 658, 625 cm-1. 1H NMR (600 MHz, DMSO-d6): δ 14.69 (1H, s, OH), 8.68 (1H, s, N=CH), 7.82–7.78 (3H, m, H2, H6, H6′), 7.65 (1H, d, J = 2.5 Hz, H4′), 7.54–7.51 (2H, m, H3, H5), 7.32 (2H, s, SO2NH2), 4.90 (2H, s, CH2). 13C NMR (151 MHz, DMSO): δ 166.75, 161.74, 143.94, 141.81, 138.30, 134.51, 128.89, 126.63, 119.32, 114.17, 107.10, 58.84. Elemental analysis for C14H12Br2N2O3S (448.13); calculated: C, 37.52; H, 2.70; N, 6.25, found C, 37.47; H, 2.67; N, 6.34.
(E)-4-{[(2-Hydroxy-3,5-diiodobenzylidene)amino]methyl}benzenesulfonamide 2i
Yellow solid; yield 95% (method A); mp 218°C. IR (ATR): 3323, 1637, 1583, 1489, 1477, 1407, 1323, 1309, 1237, 1208, 1160, 1126, 1097, 1019, 912, 868, 839, 810, 729, 681, 664, 650, 633 cm-1. 1H NMR (500 MHz, DMSO-d6): δ 14.68 (1H, s, OH), 8.63 (1H, s, N=CH), 8.05 (1H, d, J = 2.1 Hz, H6′), 7.85–7.82 (2H, m, H2, H6), 7.79 (1H, d, J = 2.1 Hz, H4′), 7.56–7.52 (2H, m, H3, H5), 7.35 (2H, s, SO2NH2), 4.92 (2H, s, CH2). 13C NMR (126 MHz, DMSO): δ 166.14, 163.95, 148.63, 143.53, 141.51, 140.88, 128.52, 126.25, 118.80, 91.74, 77.88, 58.40. Elemental analysis for C14H12I2N2O3S (542.13); calculated: C, 31.02; H, 2.23; N, 5.17, found C, 30.95; H, 2.18; N, 5.25.
(E)-4-{[(3-Bromo-5-chloro-2-hydroxybenzylidene)amino]methyl}benzenesulfonamide 2j
Yellow solid; yield 89% (method A); mp 232°C. IR (ATR): 3328, 1641, 1601, 1490, 1410, 1322, 1310, 1229, 1204, 1161, 1136, 1099, 1021, 919, 866, 839, 810, 735, 681, 662, 642, 609 cm-1. 1H NMR (600 MHz, DMSO-d6): δ 14.67 (1H, s, OH), 8.69 (1H, s, N=CH), 7.82–7.79 (2H, m, H2, H6), 7.72 (1H, d, J = 2.5 Hz, H6′), 7.55–7.51 (3H, m, H3, H5, H4′), 7.32 (2H, s, SO2NH2), 4.91 (2H, s, CH2). 13C NMR (151 MHz, DMSO): δ 166.78, 161.06, 143.92, 141.87, 135.82, 131.46, 128.88, 126.63, 120.51, 118.67, 113.60, 59.06. Elemental analysis for C14H12BrClN2O3S (403.67); calculated: C, 41.66; H, 3.00; N, 6.94, found C, 41.70; H, 3.06; N, 7.00.
(E)-4-{[(5-Chloro-2-hydroxy-3-iodobenzylidene)amino]methyl}benzenesulfonamide 2k
Yellow solid; yield 94% (method A); mp 231°C. IR (ATR): 3334, 1641, 1595, 1500, 1481, 1411, 1322, 1310, 1205, 1161, 1129, 1100, 1026, 918, 868, 839, 811, 715, 681, 658 cm-1. 1H NMR (500 MHz, DMSO-d6): δ 8.66 (1H, s, N=CH), 7.88 (1H, d, J = 2.6 Hz, H6′), 7.85–7.82 (2H, m, H2, H6), 7.58 (1H, d, J = 2.6 Hz, H4′), 7.57–7.54 (2H, m, H3, H5), 7.36 (2H, s, SO2NH2), 4.93 (2H, s, CH2). 13C NMR (126 MHz, DMSO): δ 166.27, 162.80, 143.53, 141.61, 141.08, 131.78, 128.56, 126.27, 120.87, 117.07, 90.24, 58.73. Elemental analysis for C14H12ClIN2O3S (450.67); calculated: C, 37.31; H, 2.68; N, 6.22, found C, 37.38; H, 2.72; N, 6.29.
(E)-4-({[(5-Nitrofuran-2-yl)methylene]amino}methyl)benzenesulfonamide 3a
White solid; yield 85% (method A); mp 176.3°C (decomp.). IR (ATR): 3390, 3135, 2992, 2887, 1642, 1575, 1515, 1485, 1413, 1402, 1359, 1351, 1327, 1312, 1239, 1148, 1100, 1056, 1020, 979, 969, 930, 821, 811, 740, 718, 683, 657 cm-1. 1H NMR (500 MHz, DMSO-d6): δ 8.49 (1H, s, N=CH), 7.83–7.80 (2H, m, H2, H6), 7.77 (1H, d, J = 3.9 Hz, H4′), 7.54–7.51 (2H, m, H3, H5), 7.33 (2H, s, SO2NH2), 7.31 (1H, d, J = 3.9 Hz, H3′), 4.90 (2H, s, CH2). 13C NMR (125 MHz, DMSO): δ 152.38, 152.28, 151.43, 143.09, 142.82, 128.66, 126.03, 116.61, 114.13, 63.58. Elemental analysis for C12H11N3O5S (309.30); calculated: C, 40.60; H, 3.58; N, 13.59, found C, 40.61; H, 3.63; N, 13.55.
(E)-4-({[(5-Nitrothiophen-2-yl)methylene]amino}methyl)benzenesulfonamide 3b
Brown solid; yield 65% (method A); mp 152.2°C. IR (ATR): 3295, 3104, 1634, 1535, 1509, 1443, 1409, 1340, 1314, 1286, 1244, 1212, 1187, 1159, 1150, 1122, 1100, 1034, 917, 834, 812, 732, 681, 634 cm-1. 1H NMR (600 MHz, acetone-d6): δ 8.68 (1H, s, N=CH), 8.01 (1H, d, J = 4.2 Hz, H4′), 7.86 (2H, d, J = 8.4 Hz, H2, H6), 7.55–7.50 (3H, m, H3, H5, H3′), 6.52 (2H, s, SO2NH2), 4.91 (2H, s, CH2). 13C NMR (151 MHz, acetone): δ 155.99, 155.88, 148.61, 143.30, 143.14, 129.93, 129.30, 128.38, 126.35, 63.13. Elemental analysis for C12H11N3O4S2 (325.02); calculated: C, 44.30; H, 3.41; N, 12.92, found C, C, 40.27; H, 3.49; N, 12.99.
(E)-4-{[(Furan-2-ylmethylene)amino]methyl}benzenesulfonamide 3c
Brownish solid; yield 84% (method B); mp 155.0°C. IR (ATR): 3300, 1649, 1600, 1482, 1410, 1391, 1320, 1275, 1156, 1150, 1097, 1021, 926, 884, 809, 826, 765, 681, 654 cm-1. 1H NMR (500 MHz, DMSO-d6): δ 8.34 (1H, s, N=CH), 7.84–7.78 (3H, m, H2, H6, H5′), 7.51–7.48 (2H, m, H3, H5), 7.32 (2H, s, SO2NH2), 6.97 (1H, d, J = 3.4 Hz, H3′), 6.63 (1H, dd, J = 3.4, 1.8 Hz, H4′), 4.78 (2H, s, CH2). 13C NMR (125 MHz, DMSO): δ 151.55, 151.50, 145.73, 143.78, 142.85, 128.41, 125.94, 114.96, 112.20, 63.42. Elemental analysis for C12H12N2O3S (264.30); calculated: C, 54.53; H, 4.58; N, 10.60, found C, 54.51; H, 4.60; N, 10.57.
(E)-4-{[(Thiophen-2-ylmethylene)amino]methyl}benzenesulfonamide 3d
White solid; yield 80% (method B); mp 154.7°C. IR (ATR): 3290, 2944, 1630, 1430, 1410, 1331, 1305, 1228, 1186, 1156, 1096, 1051, 1022, 851, 813, 779, 755, 723, 673 cm-1. 1H NMR (500 MHz, DMSO-d6): δ 8.61 (1H, s, N=CH), 7.78–7.75 (2H, m, H2, H6), 7.65 (1H, d, J = 5.3 Hz, H5′), 7.49 (1H, d, J = 3.6 Hz, H3′), 7.47–7.44 (2H, m, H3, H5), 7.28 (2H, s, SO2NH2), 7.13 (1H, dd, J = 5.0, 3.6 Hz, H4′), 4.75 (2H, s, CH2). 13C NMR (125 MHz, DMSO): δ 156.80, 144.13, 143.22, 142.56, 132.39, 130.40, 128.76, 128.36, 126.34, 63.19. Elemental analysis for C12H12N2O2S2 (280.36); calculated: C, 51.41; H, 4.31; N, 9.99, found C, 51.44; H, 4.30; N, 10.03.
(E)-4-{[(5-Chloro-2-methoxybenzylidene)amino]methyl}benzenesulfonamide 4a
White solid; yield 78% (method A); mp 178.8°C. IR (ATR): 3318, 2969, 2844, 1639, 1599, 1483, 1465, 1439, 1415, 1387, 1332, 1271, 1256, 1153, 1135, 1096, 1022, 1005, 917, 884, 827, 812, 708, 684, 645 cm-1. 1H NMR (600 MHz, DMSO-d6): δ 8.73 (1H, s, N=CH), 7.79–7.75 (3H, m, H2, H6, H6′), 7.49–7.45 (3H, m, H3, H5, H4′), 7.28 (2H, s, SO2NH2), 7.13 (1H, d, J = 8.8 Hz, H3′), 4.82 (2H, s, CH2), 3.84 (3H, s, CH3). 13C NMR (151 MHz, DMSO): δ 157.84, 157.02, 144.16, 143.20, 132.29, 128.73, 126.34, 126.31, 125.85, 125.30, 114.65, 63.94, 56.69. Elemental analysis for C15H15ClN2O3S (338.81); calculated: C, 53.18; H, 4.46; N, 8.27, found C, 53.15; H, 4.50; N, 8.27.
(E)-4-({[1-(5-Chloro-2-hydroxyphenyl)ethylidene]amino}methyl)benzenesulfonamide 4b
Yellow solid; yield 75% (method B); mp >250°C. IR (ATR): 3305, 1609, 1559, 1507, 1411, 1374, 1325, 1261, 1227, 1153, 1095, 1055, 1018, 928, 864, 830, 817, 767, 735, 652 cm-1. 1H NMR (500 MHz, DMSO-d6): δ 7.82–7.79 (2H, m, H2, H6), 7.70 (1H, d, J = 2.6 Hz, H6′), 7.54–7.51 (2H, m, H3, H5), 7.32 (2H, s, SO2NH2), 7.29 (1H, dd, J = 8.8, 2.6 Hz, H4′), 6.78 (1H, d, J = 8.9 Hz, H3′), 4.88 (2H, s, CH2), 2.45 (3H, s, CH3). 13C NMR (126 MHz, DMSO): δ 173.55, 162.78, 143.55, 143.25, 132.85, 128.73, 128.68, 126.61, 120.95, 120.52, 120.39, 52.80, 15.70. Elemental analysis for C15H15ClN2O3S (338.81); calculated: C, 53.18; H, 4.46; N, 8.27, found C, 53.21; H, 4.47; N, 8.29.
(E)-4-({[1-(3,5-Dichloro-2-hydroxyphenyl)ethylidene]amino}methyl)benzenesulfonamide 4c
Yellow solid; yield 80% (method B); mp 235.1°C. IR (ATR): 3338, 1616, 1596, 1532, 1509, 1418, 1367, 1321, 1311, 1226, 1154, 1098, 938, 856, 820, 809, 754, 728, 694, 636 cm-1. 1H NMR (500 MHz, DMSO-d6): δ 7.89–7.86 (2H, m, H2, H6), 7.70 (1H, d, J = 2.7 Hz, H6′), 7.61–7.57 (3H, m, H3, H5, H4′), 7.37 (2H, s, SO2NH2), 5.00 (2H, s, CH2), 2.58 (3H, s, CH3). 13C NMR (126 MHz, DMSO): δ 174.77, 163.41, 143.60, 141.06, 132.73, 128.53, 127.67, 126.39, 124.94, 117.87, 117.03, 50.37, 15.31. Elemental analysis for C15H14C12N2O3S (373.25); calculated: C, 48.27; H, 3.78; N, 7.51, found C, 48.24; H, 3.74; N, 7.49.
(E)-2,4-Diiodo-6-{[(4-sulfamoylbenzyl)imino]methyl}phenyl acetate 4d
Yellow-orange solid; yield 89% (method B); mp 212.9°C. IR (ATR): 3323, 1745, 1638, 1583, 1498, 1477, 1407, 1382, 1323, 1309, 1237, 1208, 1189, 1160, 1126, 1097, 1018, 911, 868, 839, 810, 729, 681, 664, 650 cm-1. 1H NMR (600 MHz, DMSO-d6): δ 8.59 (1H, s, N=CH), 8.02 (1H, d, J = 2.1 Hz, H6′), 7.81–7.78 (2H, m, H2, H6), 7.75 (1H, d, J = 2.1 Hz, H4′), 7.53–7.50 (2H, m, H3, H5), 7.31 (2H, s, SO2NH2), 4.88 (2H, s, CH2), 2.28 (3H, s, CH3). 13C NMR (151 MHz, DMSO): δ 168.70, 166.53, 164.32, 149.02, 143.91, 141.88, 141.27, 128.90, 126.63, 119.18, 92.09, 78.28, 58.77, 21.37. Elemental analysis for C16H14I2N2O4S (584.17); calculated: C, 32.90; H, 2.42; N, 4.80, found C, 32.94; H, 2.44; N, 4.83.
(E)-4-{[(4-Hydroxy-3,5-diiodobenzylidene)amino]methyl}benzenesulfonamide 4e
Yellow solid; yield 94% (method B); mp 214.6°C. IR (ATR): 3314, 3199, 1659, 1563, 1456, 1418, 1404, 1365, 1319, 1306, 1227, 1202, 1174, 1152, 1099, 1099, 1055, 1041, 920, 898, 877, 828, 804, 752, 693, 674 cm-1. 1H NMR (600 MHz, DMSO-d6): δ 10.56 (1H, s, OH), 8.45 (1H, s, N=CH), 8.11 (2H, s, H2′, H6′), 7.86–7.83 (2H, m, H2, H6), 7.57–7.54 (2H, m, H3, H5), 7.38 (2H, s, SO2NH2), 4.76 (2H, s, CH2). 13C NMR (151 MHz, DMSO): δ 160.09, 144.15, 142.33, 140.77, 129.81, 128.90, 126.51, 77.10, 49.07. Elemental analysis for C14H12I2N2O3S (542.13); calculated: C, 31.02; H, 2.23; N, 5.17, found C, 30.99; H, 2.22; N, 5.23.
(E)-4-{[(2-Hydroxy-3,5-diiodobenzyl)amino]methyl}benzenesulfonamide 4f
Yellowish solid; yield 89%; mp 180.8°C. IR (ATR): 3383, 3263, 1684, 1647, 1578, 1559, 1539, 1507, 1490, 1458, 1435, 1410, 1386, 1330, 1264, 1189, 1160, 1091, 1019, 999, 907, 876, 861, 847, 810, 755, 682, 659, 633 cm-1. 1H NMR (600 MHz, DMSO-d6): δ 7.79 (1H, d, J = 2.1 Hz, H4′), 7.77 (2H, d, J = 8.3 Hz, H2, H6), 7.47 (2H, d, J = 8.3 Hz, H3, H5), 7.34 (1H, d, J = 2.1 Hz, H6′), 7.29 (2H, s, SO2NH2), 3.82 (2H, s, CH2), 3.75 (2H, s, CH2). 13C NMR (151 MHz, DMSO): δ 158.17, 143.82, 143.23, 142.54, 136.86, 128.86, 126.08, 125.99, 87.61, 81.01, 50.76, 50.09. Elemental analysis for C14H14I2N2O3S (544.15); calculated: C, 30.90; H, 2.59; N, 5.15, found C, 30.87; H, 2.63; N, 5.11.
Biology
Antibacterial activity
The basic screening of antibacterial activity of 1–4 was evaluated against four Gram-positive and four Gram-negative strains of a clinical importance, namely S. aureus ATCC (American Type Culture Collection) 29213, CCM (Czech Collection of Microorganisms, Brno, Czech Republic) 4223, methicillin-resistant S. aureus ATCC 43300, CCM 4750 (MRSA), Staphylococcus epidermidis ATCC 12228, CCM 4418, Enterococcus faecalis ATCC 29212, CCM 4224; E. coli ATCC 25922, CCM 3954, Klebsiella pneumoniae ATCC 10031, CCM 4415, Acinetobacter baumannii ATCC 19606, DSM (German Collection of Microorganisms and Cell Cultures GmbH, Braunschweig, Germany) 30007 and Pseudomonas aeruginosa ATCC 27853, CCM 3955. The strains were obtained from CCM or DSM.
For extended study of antibacterial activity, clinical isolate strains from the genus Staphylococcus and Enterococcus, provided by the Department of Clinical Microbiology, University Hospital Hradec Králové, Czech Republic, were employed (Supplementary Table 1). All these strains were taxonomically classified by biochemical tests and MALDI-TOF (Microflex LH/SH MALDI-TOF, Bruker Biotyper 3.0 SW, Bruker Daltonic GmbH, Bremen, Germany) instrumentation. The antibiograms of strains were determined by the disc diffusion method according to EUCAST (The European Committee on Antimicrobial Susceptibility Testing) recommendation [Citation28]. Clinical isolate strain, VRSA, was kindly provided by the National Institute of Public Health in Prague (NIPH), Czech Republic. The list of clinical isolate strains with susceptibility profiles and other strain specifications is present in Supplementary Table 1.
The microdilution broth method was performed according to EUCAST instructions [Citation29] with slight modifications, starting from the concentration of investigated compounds being 0.97 μM. Briefly, the cultivation was done in Cation-adjusted Mueller-Hinton broth (CAMHB, M-H 2 Broth, Sigma-Aldrich, MO, USA) at 35 ± 2°C. Tested compounds were dissolved in DMSO (Sigma-Aldrich, MO, USA) to produce stock solutions. The final concentration of DMSO in the testing medium did not exceed 1% (v/v) of the total solution composition and did not affect the growth of bacteria. Positive (microbes solely), negative (cultivation medium and DMSO) controls and internal quality standards were involved in each assay. Antibacterial activity is expressed as MIC (reported in μM) after 24 and 48 h of static incubation in dark and humidified atmosphere at 35 ± 2°C. MIC was determined by naked eye in the well with the lowest drug concentration, where no visible growth of microbial agent was detected.
Parent mafenide (as acetate) 1 together with broad-spectrum beta-lactam antibiotic piperacillin (PIP) were used as reference compounds. The results of PIP were read after 24 h. Standard antibiotics (ciprofloxacin, gentamicin; data not shown) and internal quality control/reference strains are routinely included in parallel testing-basic screening of antibacterial activity.
Pharmacodynamic profiling of the in vitro interaction of 2i & 4d with selected staphylococcal strains
Two staphylococcal clinical isolate strains of the greatest clinical significance, MRSA 131/16 and VRSA (NIPH), were chosen for mapping of the impact of two compounds (2i and 4d) within at least the first 18 h of mutual interaction in vitro. The microdilution broth method mentioned earlier was employed. Each period of 30 min of incubation at 36.8°C, the optical density was measured and related to the time (Bioscreen C instrument, 540 nm, Oy Growth Curves, Finland).
Antimycobacterial activity evaluation
Antimycobacterial assay was performed with fast growing Mycobacterium (Mycolicibacterium) smegmatis ATCC 607 (DSM 43465), Mycobacterium (Mycolicibacterium) aurum ATCC 23366 (DSM 43999) from DSM (Braunschweig, Germany) and with avirulent strain of Mycobacterium tuberculosis H37Ra ATCC 9431 (ITM-M006710) from Belgian Co-ordinated Collections of Microorganisms. The technique used for activity determination was the microdilution broth panel method using 96-well microtitration plates. Culturing medium was Middlebrook 7H9 broth (Merck, Steinheim, Germany) enriched with 0.4% of glycerol (Merck) and 10% of Middlebrook OADC (Oleic Albumin Dextrose Catalase) growth supplement (Himedia, Mumbai, India).
Mycobacterial strains were cultured on Middlebrook 7H9 agar and suspensions were prepared in Middlebrook 7H9 broth. Final density was adjusted to value 1.0 according to McFarland scale and diluted in ratio 1:20 (for fast growing mycobacteria) or 1:10 (for M. tuberculosis) with broth.
Tested compounds were dissolved in DMSO (Merck, Steinheim, Germany) then broth was added to obtain concentration 2000 μg/ml. Standards used for activity determination were isoniazid (INH) and parent mafenide acetate 1. Final concentrations were reached by binary dilution and addition of mycobacterial suspension and were set at 500, 250, 125, 62.5, 31.25, 15.625, 7.81 and 3.91 μg/ml or 50, 25, 12.5, 6.25, 3.13, 1.56, 0.78 and 0.39 μg/ml according to final MIC. INH was diluted in range of 500–3.91 μg/ml for screening against fast growing mycobacteria and in range of 1–0.0078 μg/ml for screening against M. tuberculosis. The final concentration of DMSO did not exceed 2.5% (v/v) and did not affect the growth of M. smegmatis, M. aurum or M. tuberculosis. Positive (broth, DMSO, bacteria) and negative (broth, DMSO) growth controls were included.
Plates were sealed with polyester adhesive film and incubated in dark at 37°C without agitation. The addition of 0.01% solution of resazurin sodium salt followed 48 h of incubation for M. smegmatis, 72 h of incubation for M. aurum and after 120 h of incubation for M. tuberculosis, respectively. Microtitration panels were then incubated for further 2.5 h for determination of activity against M. smegmatis, 4 h for M. aurum and 24 h for M. tuberculosis, respectively.
Antimycobacterial activity was expressed as minimum inhibition concentration and the value was read on the basis of stain colour change (blue colour – active compound; pink colour – inactive compound). MIC values of INH were 15.625 μg/ml, 3.91 μg/ml and 0.25 μg/ml for M. smegmatis, M. aurum and M. tuberculosis, respectively. All experiments were performed in duplicate.
Three derivatives (the most active 3a, 3b and 2i) were evaluated against seven multidrug-resistant TB strains (dilution of these strains was 10-3) with different resistance patterns. All of them were resistant to INH, rifamycines and streptomycin including isolate 9449/2007. For some isolates, an additional resistance was present: strain 7357/1998 was also resistant to ethambutol and ofloxacin, strain 234/2005 to ethambutol, strain 8666/2010 resistant to ethambutol, ofloxacin and clofazimine, strain Praha 1 exhibited an additional resistance to ethambutol and clofazimine; Praha 4 to ethambutol, ofloxacin and clofazimine; and Praha 131 was resistant to INH, rifampicin, rifabutin, streptomycin, ethambutol, ofloxacin, gentamicin and amikacin (i.e., XDR-TB strain). The following concentrations were used: 1000, 500, 250, 125, 62.5, 32, 16, 8, 4, 2 and 1 μM.
Antifungal activity
Antifungal activity was evaluated against four yeast strains, namely Candida albicans ATCC 24443, CCM 8320, Candida krusei ATCC 6258, CCM 8271, Candida parapsilosis ATCC 22019, CCM 8260, Candida tropicalis ATCC 750, CCM 8264; and four strains of filamentous fungi, namely Aspergillus fumigatus ATCC 204305, Aspergillus flavus CCM 8363, Lichtheimia corymbifera CCM 8077 and Trichophyton interdigitale ATCC 9533, CCM 8377. A microdilution broth method was performed according to EUCAST instructions [Citation30,Citation31] with slight modifications. Briefly, tested compounds were dissolved in DMSO and diluted in a twofold manner with RPMI-1640 medium with L-glutamine, supplemented with 2% glucose (w/v) and buffered to pH 7.0 with 3-(N-morpholino)propane-1-sulfonic acid (all components were purchased from Sigma-Aldrich, MO, USA). The final concentration of DMSO in the tested medium did not exceed 1% (v/v) of the total solution composition, and this concentration did not inhibit the fungal growth. Static incubation was performed in dark and humidified atmosphere, at 35 ± 2°C for 24 and 48 h (72 and 120 h for Trichophyton interdigitale). Positive controls consisted of test fungus solely, while negative controls consisted of medium and DMSO. Visual inspection was used for MIC endpoint evaluation. MIC was determined by the naked eye in the well with the lowest drug concentration, where no visible growth of microbial agent was detected. MIC determination scale started from 0.97 μM.
Parent mafenide acetate 1 and antimycotic drug fluconazole were involved as reference compounds for a comparison. MIC values of fluconazole mean IC50 values, i.e., the lowest drug concentration giving growth inhibition of 50% of that of the drug-free control. Results were read after 24 h (yeasts) or 48 h (molds) microdilution plates cultivation without agitation at 35 ± 2°C in humidified atmosphere. The results were read with a microdilution plate reader (SynergyTM HTX, BioTek Instruments, Inc., VT, USA) at wavelength 530 nm.
Internal quality control was included too. Standard antimycotics (voriconazole, amphotericin B; data not shown) are routinely included in parallel testing-basic screening of antibacterial activity.
Cytotoxicity determination
The human hepatocellular liver carcinoma cell line HepG2 (passage 18-19) purchased from Health Protection Agency Culture Collections (ECACC, Salisbury, UK) was cultured in Minimum Essentials Eagle Medium supplemented with 10% fetal bovine serum, 1% L-glutamine solution and 1% non-essential amino acid solution in a humidified atmosphere containing 5% CO2 at 37°C. For subculturing, the cells were harvested after trypsin/EDTA treatment at 37°C. To determine cytotoxicity of the compounds, the cells treated with the tested substances were used as experimental groups whereas untreated cells served as control groups. All used chemicals were obtained from Merck (MO, USA).
The cells were seeded in a density of 15,000 cells per well in a 96-well plate. The next day the cells were treated with each of the tested substances at a broad range of concentrations (based on their solubility) in triplicates. The compounds were dissolved in DMSO (maximal incubation concentration of DMSO was 1% v/v). The controls representing 100% cell viability, 0% cell viability (treated with 10% DMSO), no cell control and vehiculum control were incubated in parallel also in triplicates. After 24 h of incubation in a humidified atmosphere containing 5% CO2 at 37°C, the reagent from the kit CellTiter 96 Aqueous One Solution Cell Proliferation Assay (CellTiter 96; PROMEGA, WI, USA) was added. After 2 h of incubation at 37°C, absorbance of samples was recorded at 490 nm (TECAN, Infinita M200, Grödig, Austria). A standard toxicological parameter IC50 was calculated by nonlinear regression from a semilogarithmic plot of incubation concentration versus percentage of viability relative to untreated controls using GraphPad Prism 8 software (GraphPad Software, Inc., CA, USA).
Results of the experiments are presented as inhibitory concentrations that reduce viability of the cell population to 50% from the maximal viability (IC50). Known cytotoxic compounds tamoxifen (as tamoxifen citrate) and cisplatin were involved as reference compounds.
Results & discussion
Design & synthesis
To modify MAF, we originally used halogen-substituted salicylaldehydes to increase lipophilicity according to our previous publications [Citation23,Citation24,Citation32] together with 5-nitrofurfural and 5-nitrothiophene-2-carbaldehyde, known pharmacophores for antimicrobial activity [Citation33,Citation34] (derivatives 2a-2k; 3a and 3b). Once we had results of their biological activity in hand, we designed and synthesized additional analogs to investigate structure–activity relationships (SAR; Supplementary Figure 1). These modifications involved the removal of nitro group from heterocyclic derivatives (3c, 3d), replacement of imine hydrogen with methyl (ketone-based derivatives 4b and 4c), etherification and esterification of the phenolic hydroxyl (4a, 4d, respectively), positional isomer 4e and reduction of the double bond (4f). The synthesis of the target compounds is shown in .
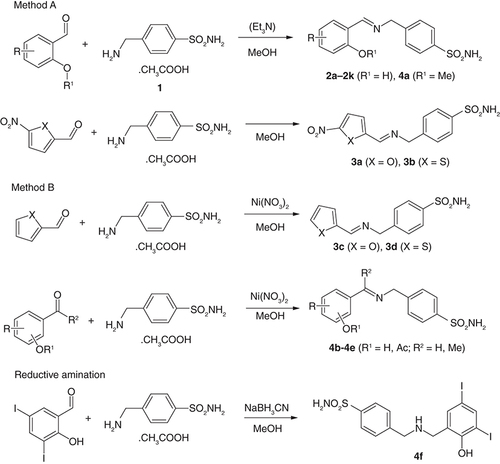
MAF-based imines were synthesized from MFA and aldehydes/ketones using two methods. Method A consists of the reaction of the sulfonamide with the corresponding aldehydes (1.1 eq.; salicylaldehydes, 2a-2k, 5-nitrofuran/thiophen-2-carbaldehydes 3a and 3b and 5-chloro-2-methoxybenzaldehyde, 4a) in boiling methanol for 3 h. Method B involves adding nickel(II) nitrate hexahydrate as a catalyst (0.1 eq.) to methanolic solution of MAF and carbonyl compounds (1.1 eq.) and stirring at room temperature for 12 h. It was used for thiophene/furan-2-carbaldehydes (3c, 3d), ketones (4b, 4c), ester (4d) and 4-hydroxy-3,5-diiodobenzaldehyde derivative 4e. In the method A, an addition of base (1 equiv. of triethylamine) has no effect on the yields. In some cases, spontaneous precipitation of imine was not observed, so the reaction mixture was diluted with water to initiate crystallization. Very good yields were within the range of 65–95%. Method B gave high yields of 75–94%, too. For the synthesis of ester-based imine 4d, 2-formyl-4,6-diiodophenyl acetate was synthesized from 3,5-diiodosalicylaldehyde by treatment with a mild excess of triethylamine and acetyl chloride.
The reduced analog 4f was prepared by reductive amination using sodium cyanoborohydride in methanol (yield of 89%; ).
Representative spectra are given in the Supplementary Material.
Antibacterial activity
Based on the clinical use of MAF, we primarily evaluated the antibacterial activity of compounds 2-4. The panel of human pathogens includes four Gram-positive bacterial strains (S. aureus ATCC [American Type Culture Collection] 29213 [methicillin-susceptible, MSSA], methicillin-resistant S. aureus [MRSA] ATCC 43300, Staphylococcus epidermidis ATCC 12228, Enterococcus faecalis ATCC 29212; ) and four Gram-negative strains (E. coli ATCC 25922, Klebsiella pneumoniae ATCC 10031, Acinetobacter baumannii ATCC 19606 and Pseudomonas aeruginosa ATCC 27853; Supplementary Table 2). Piperacillin and parent MFA 1 were used as standards for comparison.
Table 1. Structures and activity against Gram-positive bacteria and cytotoxicity of mafenides 1–4.
The derivatives showed MIC starting at 7.81 μM (2i) and only four of them were completely inactive against all included strains at 500 μM (2c, 3d, 4b and 4f; ). Three staphylococcal strains have a similar susceptibility profile to our derivatives, with S. epidermidis being slightly more sensitive. Importantly, there is no cross-resistance to β-lactams as evidenced by identical MIC values of methicillin-resistant and susceptible S. aureus (MIC of ≥7.81 μM). E. faecalis was inhibited at higher concentrations (from 62.5 μM for diiodinated Schiff bases 2i and 4d).
Among salicylidene imines, the lowest MIC values are associated with the presence of 3,5-I2 (2i and its acetyl ester 4d), and other halogens or monohalogenation provided milder activity. Replacement of ‘imine' hydrogen by methyl (4b and 4c) is detrimental, as is positional isomer of phenol group (4e) and reduction of double bond (4f). Comparing heteroarylidene imines 3 to each other, a 5-NO2 group is necessary (3a vs. 3c, 3b vs. 3d) and thiophene provided lower MIC than isosteric furan (3b vs. 3a, respectively). The Schiff base 3b is one of the most potent antibacterial agents in this study.
MIC of mafenide (as salt with acetic acid) exceeded 500 μM and, fortunately, all its active imines represent more potent and successful modifications. Schiff bases inhibited β-lactamase producing piperacillin-resistant MRSA strain, but this β-lactam drug remains superior against the remaining susceptible strains.
Keeping these promising results in mind, we subjected five highly potent imines (2i, 2k, 3a, 3b and 4d) for advanced antibacterial evaluation against a panel of drug-resistant Gram-positive bacteria: S. aureus including MRSA and a vancomycin-resistant strain (VRSA), S. epidermidis, Staphylococcus lugdunensis and vancomycin-resistant enterococci (VRE) covering both collection strains and clinical isolates. Their susceptibility profiles are summarized in supplementary material (Supplementary Table 1). The results are summarized in . Optical density measurements and visual inspection were used in evaluation.
Table 2. Activity of selected mafenide derivatives 2–4 against drug-resistant Gram-positive cocci.
All the five imines retained antimicrobial activity even against drug-resistant Gram-positive strains. Notably, 5-nitrofurylidene, 3-I-5-Cl and acetylated 3,5-diiodo Schiff bases (3a, 2k and 4d, respectively) exhibited identical MIC values as for susceptible Gram-positives consistently, whereas the remaining two molecules were found similarly active only against some species. Fortunately, the most clinically relevant multiresistant superbugs were inhibited by two imines (2i and 4d) including polyresistant VRE strains 17/21 and 18/21 that share the lowest susceptibility.
The bactericidal action can be partially indicated by MIC values ( & ) evaluated after 24 and 48 h of incubation. The recognition of no shift or one-step shift in MIC values (i.e., ± one dilution in the range of concentration with serial dilution) corresponding to one compound acting on one selected strain, allows a preliminary estimation of the bactericidal effect (salicylic derivatives 2i, 2k and 4d and thiophene 3b). This prediction approach is based on the fact that bactericidal compounds significantly reduce the number of viable cells (≥99.9% reduction of viability). Then, an additional 24 h period is sufficient to increase the number of surviving cells to a detectable level. In contrast, 5-nitrofurylidene-mafenide conjugate 3a was most likely bacteriostatic with an increase of MIC values.
Altogether, these compounds have a great potential against Gram-positive cocci, including resistant strains, regardless of the resistance profile.
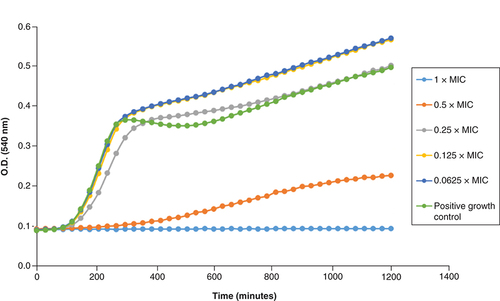
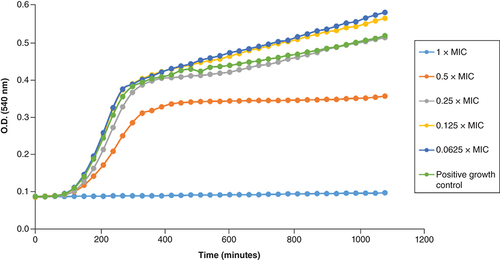
We further investigated the time-dependent interactions of 2i and 4d with a collection strain of VRSA (NIPH) and a polyresistant MRSA 131/16 at various concentrations related to their MIC values. We used four sub-MIC concentrations (0.0625, 0.125, 0.25 and 0.5×MIC) and 1×MIC. The dependence of bacterial density on time and inhibitor concentration is shown in & & Supplementary Figures 2 & 3. The three lowest sub-inhibitory concentrations of 2i (0.0625–0.25×MIC) did not lead to suppression of bacterial cell biomass formation in both strains ( & ). In addition, concentrations of 0.0625 and 0.125×MIC induced slightly higher biomass formation. This can perhaps be explained on the basis of bacterial behavior under the influence of a bactericidal antibiotic. The cidal ones can cause in the sub-inhibitory concentrations a strong stress response, which is manifested by an increase in metabolic activity. The aim of its increase is primarily the repair and the formation of new cells. On the other hand, it is important that at a concentration of 0.5×MIC, only limited bacterial growth was observed at the same time. Interestingly, this phenomenon was not observed for 4d (Supplementary Figures 2 & 3), where all sub-MIC concentrations caused a decrease in bacterial density, although for the two lowest concentrations only after a prolonged period of time for MRSA.
MIC: Minimum inhibitory concentration; VRSA: Vancomycin-resistant S. aureus.
MIC: Minimum inhibitory concentration; MRSA: Methicillin-resistant S. aureus.
Focusing on Gram-negatives, the activity was slightly lower, and some compounds (not listed in Supplementary Table 2) were inactive. 3,5-Diiodo and thiophene derivatives (2i and 3b, respectively) showed the best efficacy with MIC starting from 15.62 μM. These two imines inhibited all four species in the concentration range used. Notably, 2i was superior to standard β-lactam piperacillin against K. pneumoniae and 3b for A. baumannii. In addition, all active Schiff bases enhanced activity of parent MFA that is mainly used to treat infections caused by Gram-negative pathogens. However, its MIC values are higher than those used in our concentration range. Glasser et al. [Citation7] reported MIC of MFA against both Gram-positive and Gram-negative strains in the range of 128–2048 μM.
Sulfonamides have also been reported as antimycobacterial agents to combat both Mycobacterium tuberculosis (Mtb.) and non-tuberculous mycobacteria (NTM) [Citation25,32]. Hence, mafenide derivatives 2-4 were also evaluated in vitro against three mycobacteria: Mtb. H37Ra ITM-M006710 and two NTM – Mycobacterium (Mycolicibacterium) smegmatis DSM (German Collection of Microorganisms and Cell Cultures GmbH, Braunschweig, Germany) 43465 and Mycobacterium (Mycolicibacterium) aurum DSM 43999. Isoniazid (INH), a first line antituberculosis drug, and the parent MFA 1 were used to compare MIC values ().
Table 3. Antimycobacterial activity of the mafenides 1–4.
The compounds produced MIC values ranging from 3.91 μg/ml (3b), two compounds (imine 4b and reduced analog 4f) were inactive (MIC of ≥500 μg/ml). The best inhibition was associated with the presence of 5-nitrofurylidene and especially (5-nitrothiophen-2-yl)methylidene moieties (3a and 3b). Removal of NO2 group resulted in a decrease in activity (up to two orders of magnitude; 3c and 3d); this substitution pattern is crucial. Among salicylic compounds, 3,5-dihalogenosalicylidenes with heavier halogens than fluorine have the highest antimycobacterial activity (2g-2k). Substitution of imine double bond with methyl (i.e., acetophenone derivatives) resulted in higher MIC (2c vs. 4b; 2g vs. 4c). Interestingly, salicylic 2-OH group is not essential, because its methylation (4a), acetylation (4d) and change of position (4e) are tolerated.
M. smegmatis was slightly less susceptible, whereas results for more sensitive Mtb. and M. aurum were comparable. The most potent nitro compounds 3a and 3b produced identical activity to INH in M. aurum and M. smegmatis, but not in Mtb., which is very sensitive to this drug. Parent mafenide acetate 1 showed mild antimycobacterial properties (MIC from 250 μg/ml) and, importantly, almost all imines share significantly improved inhibition of mycobacteria (up to 64× for 3b). Obviously, this modification can be considered as very successful.
Three imines with the highest in vitro efficacy against mycobacteria (i.e., 3a, 3b and 2i as a representative of halogenated salicylidene derivatives) were also evaluated against five multidrug-resistant and one extensively drug-resistant TB strains with various resistance profile (Supplementary Table 3).
Importantly, all the compounds retained antitubercular activity despite resistance profile. This indicates no cross-resistance to established anti-TB drugs. Reflecting activity against NTM and susceptible Mtb. strains, the lowest MIC were found for thiophene derivative 3b (16–32 μM), followed by 5-nitrofurylidene imine 3a (16–64 μM), while salicylidene analog 2i was less potent in vitro (125–250 μM).
With respect to these results, MAF-derived imines seem to be broad-spectrum antimycobacterial agents with a different mechanism of action than clinically used drugs (INH, rifamycines, streptomycin, ethambutol, clofazimine, fluoroquinolones and aminoglycosides).
Antifungal activity
Barsoumian et al. [Citation10] found antifungal activity of MAF against clinical isolates of various molds. Prolonged exposure and high concentrations were required for growth inhibition. Therefore, in view of this study, we also determined the antifungal potential of the derivatives 2-4. Together with established antimycotic drug fluconazole and MFA, they were tested for eight human fungal pathogens: Candida albicans ATCC 24443, Candida krusei ATCC 6258, Candida parapsilosis ATCC 22019 and Candida tropicalis ATCC 750 (yeasts); Aspergillus fumigatus ATCC 204305, Aspergillus flavus CCM 8363, Lichtheimia corymbifera CCM 8077 and Trichophyton interdigitale ATCC 9533 (molds; ).
Table 4. Antifungal activity of selected mafenide derivatives 1–4.
MFA 1 had no antifungal effect under our assay conditions (MIC >500 μM), as did some derivatives (not listed in ; 2a, 2c, 3c, 3d, 4a-4c, 4e and 4f). From this point of view, the basic structural requirements for antifungal activity are as follows: (1) presence of salicylic 2-hydroxy group, it can only be esterified but not etherified (2i vs. 4d/4a and 4e); (2) aldehyde-derived imine bond that cannot be reduced (2i vs. 4f) or substituted with methyl (4b, 4c); (3) 5-nitro group on five-membered heterocycles, its removal abolishes activity (3a and 3b vs 3c and 3d).
L. corymbifera was the most resistant strain (MIC of ≥500 μM), followed by aspergilli. T. interdigitale and C. albicans showed the highest susceptibility among molds and yeasts (MIC from 15.62 and 3.9 μM, respectively).
For antifungal activity, 3,5-diiodosalicylidene scaffold (2i) is associated with high efficacy in vitro. Phenol can be acetylated (4d). In particular, other imines based on 3,5-dihalogenosalicylaldehydes containing at least one heavier halogen (Br, I; 2h, 2j and 2k) showed a significant and broad-spectrum antifungal action. Comparing five-membered rings that are more active against yeasts of the genus Candida, thiophene is preferred over furan (3a vs 3b).
MFA 1 avoided any activity up to the concentration of 500 μM, and most of its imines provided a mild-to-excellent improvement in antifungal potency (up to ≥128× for 2i). Interesting results were found when drawing a comparison of MIC values of Schiff bases to fluconazole. Our derivatives inhibited fluconazole-resistant species (C. krusei, A. flavus, A. fumigatus, L. corymbifera). In C. albicans, three iodinated derivatives (2i, 2k and 4d) showed similar MIC values to fluconazole. Four compounds were also comparable or even superior at inhibiting T. interdigitale (2e, 2h, 2i and 4d). MIC for fluconazole means 50% inhibition of growth (i.e., IC50), while it means complete inhibition for our imines. Thus, even with similar numerical values, the derivatives offered better antifungal properties.
Cytotoxicity evaluation
Cytotoxicity of the Schiff bases, their analogs and parent MFA was determined using standard hepatic cell line HepG2 and commercially available colorimetric-based CellTiter 96 assay. It is based on the reduction of tetrazolium dye to formazan in living cells, which is then detected. The amount of formazan depends on the availability of NAD(P)H in living cells.
The results of cytotoxicity evaluation of 1–4 quantified as IC50 are summarized in .
The parent mafenide acetate was nontoxic at the concentration of 500 μM, as were imines derived from unsubstituted salicylaldehyde (2a), fluorinated salicylaldehydes (2b, 2f), 5-chlorosalicylaldehyde (2c), its methyl analogs (O-methyl 4a and acetophenone 4b) and heterocyclic aldehydes 3c and 3d. Contrarily, 5-nitro analogs of the previously mentioned aldehydes yielded the most cytotoxic compounds with IC50 of 54.38 and 71.93 μM for thiophene (3b) and furan (3a) analogs, respectively. Thus, an introduction of the nitro group and the presence of sulfur potentiate toxicity to eukaryotic cells. The last group of compounds contains remaining derivatives with a relatively low toxicity with IC50 of 103.1–470.3 μM. Generally, polyhalogenation and the presence of heavier halogens resulted in enhanced cytotoxicity for HepG2. Reduction of the double bond or acetylation of 2i has no significant influence on this activity.
No potent and selective cytotoxic agent was identified. Some compounds showed a sufficient selectivity for specific antimicrobial activities (e.g., 2h–2k and 4d). From an alternative point of view, several derivatives can be considered as dual cytotoxic and broad-spectrum antimicrobial agents, in particular 3a and 3b. In contrast to traditional view, this coincidence is a quite common feature. For example, antibacterial fluoroquinolones have exhibited multitargeting anticancer properties [Citation35] and anticancer drug tamoxifen has been described as an antibacterial [Citation36] and antifungal [Citation37] agent. Their derivatives have also been investigated for this dual action [Citation38,Citation39].
The combined antimicrobial and anticancer activity can be considered as promising for many reasons. The number of patients suffering from cancer and multidrug-resistant infections is increasing, and many people are experiencing their coincidence. Chronic infections, specifically chronic inflammation or a weakened immune system, contribute to one of the important causes of cancer. Similarly, immunosuppression associated with cancer and/or its therapy paves the way for opportunistic infections [Citation40]. Although the treatment of both cancer and serious infections in immunocompromised patients is usually multimodal, the dual activity targeting this junction offers a potential advantage.
Conclusion
Simple chemical modification of established drugs can provide derivatives with improved activity profile when compared to the original. This advantage can be further enhanced by a combination of two bioactive scaffolds for designing mutual or hybrid (pro)drugs.
Mafenide, as a sulfonamide analog with a different mechanism of action and no cross-resistance, is used for the treatment of local infections. We prepared imines of mafenide and substituted salicylaldehydes or furan/thiophen-2-carbaldehydes. Based on biological results, their additional analogs were synthesized. Schiff bases exhibited potent broad-spectrum activity against Gram-positive pathogens including multidrug-resistant isolates, Gram-negative bacteria, mycobacteria as well as fungi with MIC values starting from 3.9 μM. The conversion of the parent mafenide to its imines led mainly to increased activity and bactericidal effect. Several derivatives were selective for antimicrobial action, while others showed potentially advantageous dual antimicrobial and cytotoxic activity against cancer cell line. Key structure–activity relationships were also identified.
Future perspective
Mafenide, an old and simple antibacterial drug, has key advantages, including activity against some clinically problematic pathogens, no cross-resistance to chemically similar sulfonamides and relatively low population exposure in recent years. On the other hand, it has low milligram potency, only bacteriostatic action and lower lipophilicity. In the near future, mafenide and other sulfonamide scaffolds will be modified to diminish these drawbacks, such as lower MIC, expand the number of susceptible pathogens, change the activity into bactericidal, and synthesize more lipophilic derivatives to improve the crossing of biological barriers. Of particular interest will be to target the strains that are resistant to widely used antimicrobial drugs. Mutual, hybrid compounds combining two antimicrobial scaffolds with different mechanisms of action will be beneficial to prevent the emergence of acquired drug resistance and to potentiate each other's effect.
Schiff bases combining two antimicrobial scaffolds, mafenide and salicylaldehydes, furan- or thiophen-2-carbaldehydes, were prepared in high yields to improve pharmacological profile.
These hybrid compounds exhibited potent activity against Gram-positive and Gram-negative bacteria.
The most potent imines showed activity against drug-resistant clinical isolates of Gram-positive cocci including methicillin- and vancomycin-resistant staphylococci and enterococci with no cross-resistance.
Mafenide-derived imines also inhibited human pathogenic fungi (molds and yeasts).
Mafenide-based compounds also demonstrated excellent activity against various mycobacterial strains including multidrug-resistant Mycobacterium tuberculosis.
The most effective antimicrobial agents were compounds derived from iodinated 3,5-disubstituted salicylaldehydes and 5-nitrofuran/thiophene-2-carbaldehydes.
In contrast to bacteriostatic mafenide, the results indicate a bactericidal effect on Gram-positive cocci.
Some derivatives share an acceptable selectivity for the inhibition of bacteria, while some of them exhibited potentially beneficial dual antimicrobial and cytotoxic properties.
Supplemental Text 1
Download TIFF Image (2.4 MB)Supplemental Text 1
Download TIFF Image (2.4 MB)Supplemental Text 1
Download TIFF Image (2.4 MB)Supplemental Text 1
Download TIFF Image (2.4 MB)Supplemental Text 1
Download TIFF Image (2.4 MB)Supplemental Text 1
Download TIFF Image (2.4 MB)Supplemental Text 1
Download TIFF Image (2.4 MB)Supplemental Text 1
Download TIFF Image (2.4 MB)Supplemental Text 1
Download TIFF Image (2.4 MB)Supplemental Text 1
Download TIFF Image (2.4 MB)Supplemental Text 1
Download TIFF Image (2.4 MB)Supplemental Text 1
Download TIFF Image (2.4 MB)Supplemental Text 1
Download TIFF Image (96.6 KB)Supplemental Text 1
Download TIFF Image (117.6 KB)Supplemental Text 1
Download TIFF Image (103.5 KB)Supplemental Text 1
Download MS Word (15.2 KB)Supplemental Text 1
Download MS Word (15.6 KB)Supplemental Text 1
Download MS Word (14.2 KB)Acknowledgments
The authors would like to thank I Dufková and J Vacková for their technical support and V Jakubů for providing vancomycin-resistant clinical isolate strain.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at:www.tandfonline.com/doi/full/10.2217/epi-2016-0184
Financial & competing interests disclosure
This work was supported by the project National Institute of Virology and Bacteriology [Programme EXCELES, ID Project No. LX22NPO5103 – Funded by the European Union – Next Generation EU]; Ministry of Health of the Czech Republic [grant nr. NU21-05-00482]; Czech Science Foundation [grant number 20-19638Y]; and the Charles University [SVV 260 547]. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Related Research Data
References
- Hamad A , KhanMA, RahmanKMet al. Development of sulfonamide-based Schiff bases targeting urease inhibition: synthesis, characterization, inhibitory activity assessment, molecular docking and ADME studies. Bioorg. Chem.102, 104057 (2020).
- Werth BJ . Sulfonamides. https://www.msdmanuals.com/professional/infectious-diseases/bacteria-and-antibacterial-drugs/sulfonamides
- Moore ML , MillerCS, MillerE. Substituted sulfanilamides: III. N4-acyl-N1-hydroxy derivatives. J. Am. Chem. Soc.62(8), 2097–2099 (1940).
- Cancio LC . Topical antimicrobial agents for burn wound care: history and current status. Surg. Infect.22(1), 3–11 (2021).
- Mendelson JA , LindseyD. Sulfamylon (mafenide) and penicillin as expedient treatment of experimental massive open wounds with C. perfringens infection. J. Trauma.2, 239–261 (1962).
- Hashmi DL , HaithL. The current state of topical burn treatments: a review. Curr. Trauma Rep.5, 160–168 (2019).
- Glasser JS , GuymonCH, MendeK, WolfSE, HospenthalDR, MurrayCK. Activity of topical antimicrobial agents against multidrug-resistant bacteria recovered from burn patients. Burns36, 1172–1184 (2010).
- Afshari A , NguyenL, KahnSAet al. The effective duration of antimicrobial activity of mafenide acetate after reconstitution. J. Burn Care Res.39, 736–738 (2018).
- Han CY , YangY, YuAQet al. Investigation on the mechanism of mafenide in inhibiting pyroptosis and the release of inflammatory factors. Eur. J. Pharm. Sci.147, 105303 (2020).
- Barsoumian A , SanchezCJ, MendeKet al. In vitro toxicity and activity of Dakin's solution, Mafenide acetate, and amphotericin B on filamentous fungi and human cells. J. Orthop. Trauma27, 428–436 (2013).
- Eagon RG , McManusAT. The effect of mafenide on dihydropteroate synthase. J. Antimicrob. Chemother.25, 25–29 (1990).
- U.S. Food and Drug Administration. SULFAMYLON (Mafenide Acetate, USP) ® FOR 5% TOPICAL SOLUTION. https://www.accessdata.fda.gov/drugsatfda_docs/label/1998/19832lbl.pdf.
- Zhang XJ , HeggersJP, ChinkesDL, WolfSE, HawkinsHK, WolfeRR. Topical sulfamylon cream inhibits DNA and protein synthesis in the skin donor site wound. Surgery139(5), 633–639 (2006).
- Punjataewakupt A , NapavichayanunS, AramwitP. The downside of antimicrobial agents for wound healing. Eur. J. Clin. Microbiol. Infect. Dis.38, 39–54 (2019).
- Capasso C , SupuranCT. Inhibition of bacterial carbonic anhydrases as a novel approach to escape drug resistance. Curr. Top. Med. Chem.17(11), 1237–1248 (2017).
- Selvaraj S , KaruppaiahA, KarthikS, SankarV. Synthesis and toxicity assessment of copper-based nano composite cream: an approach to enhance the antibacterial effect of mafenide acetate. Inorg. Nano-Met. Chem.51(1), 27–37 (2021).
- Harriott MM , BhindiN, KassisSet al. Comparative antimicrobial activity of commercial wound care solutions on bacterial and fungal biofilms. Anna Plast. Surg.83(4), 404–410 (2019).
- Siuda JF , CihonskiCD. New compounds – carbamate derivatives of mafenide (homosulfanilamide). J. Pharm. Sci.61(11), 1856–1857 (1972).
- Mermer A , DemirbasN, UsluH, DemirbasA, CeylanS, SirinX. Synthesis of novel Schiff bases using green chemistry techniques; antimicrobial, antioxidant, antiurease activity screening and molecular docking studies. J. Mol. Struct.1181, 151161 (2019).
- Ceramella J , IacopettaD, CatalanoA, CirilloF, LappanoR, SinicropiMS. A review on the antimicrobial activity of Schiff bases: data collection and recent studies. Antibiotics11(2), 191 (2022).
- Cebeci YU , BayrakH, ŞirinY. Synthesis of novel Schiff bases and azol-β-lactam derivatives starting from morpholine and thiomorpholine and investigation of their antitubercular, antiurease activity, acethylcolinesterase inhibition effect and antioxidant capacity. Bioorg. Chem.88, 102928 (2019).
- Kurt BZ , SonmezF, OzturkD, AkdemirA, AngeliA, SupuranCT. Synthesis of coumarin-sulfonamide derivatives and determination of their cytotoxicity, carbonic anhydrase inhibitory and molecular docking studies. Eur. J. Med. Chem.183, 111702 (2019).
- Krátký M , KonečnáK, JanoušekJet al. 4-Aminobenzoic acid derivatives: converting folate precursor to antimicrobial and cytotoxic agents. Biomolecules10, 1–21 (2020).
- Krátký M , DzurkováM, JanoušekJet al. Sulfadiazine salicylaldehyde-based schiff bases: synthesis, antimicrobial activity and cytotoxicity. Molecules22, 1573 (2017).
- Krátký M , VinšováJ, VolkováM, BuchtaV, TrejtnarF, StolaříkováJ. Antimicrobial activity of sulfonamides containing 5-chloro-2-hydroxybenzaldehyde and 5-chloro-2-hydroxybenzoic acid scaffold. Eur. J. Med. Chem.50, 433–440 (2012).
- Winthrop Chem. Co . KlarerJ. Benzenesulphonamide compounds. US2288531, 1940, A (1942).
- Tsukamoto T , YuhiK. Studies on halogenosalicylaldehyde. I: halogenosalicylaldehyde as reagent for primary amines. Yakugaku Zasshi78(7), 706–709 (1958).
- Matuschek E , BrownDF, KahlmeterG. Development of the EUCAST disk diffusion antimicrobial susceptibility testing method and its implementation in routine microbiology laboratories. Clin. Microbiol. Infect.20(4), O255–O266 (2014).
- European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID). EUCAST DISCUSSION DOCUMENT E.Dis 5.1. Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by broth dilution. Clin. Microbiol. Infect.9, 1–7 (2003).
- European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID). EUCAST DEFINITIVE DOCUMENT EDEF 7.3.1. Method for the determination of broth dilution minimum inhibitory concentrations of antifungal agents for yeasts. http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/AFST/Files/EUCAST_E_Def_7_3_1_Yeast_testing__definitive.pdf
- European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID). EUCAST DEFINITIVE DOCUMENT E.DEF 9.3.1. Method for the determination of broth dilution minimum inhibitory concentrations of antifungal agents for conidia forming moulds. http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/AFST/Files/EUCAST_E_Def_9_3_1_Mould_testing__definitive.pdf
- Krátký M , KonečnáK, JanoušekJet al. Sulfonamide-salicylaldehyde imines active against methicillin- and trimethoprim/sulfonamide-resistant Staphylococci. Future Med. Chem.13, 1945–1962 (2021).
- de Araújo Neto LN , doCarmo Alves de Lima M, de OliveiraJFet al. Synthesis, cytotoxicity and antifungal activity of 5-nitro-thiophene-thiosemicarbazones derivatives. Chem. Biol. Interact.272, 172–181 (2017).
- Krátký M , VinšováJ. Sulphur-containing heterocycles as antimycobacterial agents: recent advances in thiophene and thiadiazole derivatives. Curr. Top. Med. Chem.16(26), 2921–2952 (2016).
- Yadav V , TalwarP. Repositioning of fluoroquinolones from antibiotic to anti-cancer agents: an underestimated truth. Biomed. Pharmacother.111, 934–946 (2019).
- Al-Janabi AAHS . Repurposing of tamoxifen against the oral bacteria. Turk. J. Pharm. Sci.18, 68–74 (2021).
- Dolan K , MontgomeryS, BuchheitB, DidoneL, WellingtonM, KrysanDJ. Antifungal activity of tamoxifen: in vitro and in vivo activities and mechanistic characterization. Antimicrob. Agents Chemother.53(8), 3337–3346 (2009).
- Szczupak L , KowalczykA, TrzybińskiDet al. Organometallic ciprofloxacin conjugates with dual action: synthesis, characterization, and antimicrobial and cytotoxicity studies. Dalton Trans.49, 1403–1415 (2020).
- Abdmouleh F , ElArbi M, BenSaad Het al. Antimicrobial, antitumor and side effects assessment of a newly synthesized tamoxifen analog. Curr. Top. Med. Chem.20(25), 2281–2288 (2020).
- Felício MR , SilvaON, GonçalvesS, SantosNC, FrancoOL. Peptides with dual antimicrobial and anticancer activities. Front. Chem.5, 5 (2017).