In recent years numerous articles have been written about the abysmal productivity of the pharmaceutical industry in producing novel drugs and the enormous costs put upon the sponsors of clinical trials. The overall expenditure for innovative drug R&D is directly associated with the excessive drug candidate erosion in preclinical and clinical trials Citation[1]. Even slight improvements in timeline and attrition rates within the developmental processes would have given many patients a better quality of life and longer life spans, and most certainly would have created many more jobs in the industry Citation[2]. Analysis of the reasons for the failure resulted in the development of many new and valuable in vitro assays that allowed the early prediction of absorption, distribution, metabolism and excretion properties. For instance, drug failures as a result of poor pharmacokinetic properties or permeability were reduced dramatically from 40 to less than 20% Citation[1]. Numerous other in silico and in vitro methods are used to predict the properties of compounds and compound series. Nevertheless, the early prediction of drug toxicities that would have resulted in lower attrition rates in clinical trials has been unsuccessful, and clinical toxicity remains the main reason why drugs are failing. Since clinical toxicities of drugs have a major impact on society, the regulatory hurdles within the approval process for new medications have been enhanced. It was estimated that in the USA, 2.2 million serious adverse drug events (ADEs) occurred in a single year in hospitalized patients Citation[3]. The number of incidences for ambulant patients is unknown while 300,000 ADEs in nursing homes were registered Citation[4]. This demonstrates that ADEs are a leading cause of patient death and it was estimated that this resulted in over 100,000 fatalities in the USA per year Citation[3]. A search has begun to predict ADEs in patients early enough to terminate projects at preclinical stages. The aim is to save time and money for other projects or compound series that do not contain structural features and that cause protein interactions leading to ADEs. Specifically, care needs to be taken in the prediction of idiosyncratic toxicities. These, by definition, vary from patient to patient, occur unexpectedly and depend on the patient’s genetic background, physical state, age, gender, co-morbidities, nutritional state and immune system Citation[5]. The absence of in vivo models impairs the prediction of idiosyncratic toxicities Citation[6,7]. In addition, it could be demonstrated that many rodent models fail to predict human toxicities to an extent required to make meaningful predictions for clinical outcomes.
The current situation
Numerous in vitro toxicity assays are used routinely to guide medicinal chemists towards the best candidates in a compound series during the discovery phase. The closer the in vivo situation is replicated in any given in vitro assay for the prediction of toxicological risks, the more likely it will be to predict the outcome. For example, P450 inhibition or induction is measured frequently, allowing for the prediction of drug–drug interactions. The Ames test (bacterial reverse mutation) and RADAR screens are valuable tools to predict genotoxicity. Cytotoxicity is measured indirectly by evaluating cell membrane integrity, cell metabolism and can be assessed in a tissue-specific manner. For instance, cardiotoxicity at early discovery stages will be evaluated by HERG-channel inhibition before using any obligatory animal models. Earlier the Ashby-Tennant model was created as a warning tool for medicinal chemists Citation[8]. This model tries to correlate residues of molecules to observed carcinogenicity in rodents. In recent years efforts were undertaken to validate these empirical rules. In contrast to the model, toxicities strongly depended on the overall structure of the molecule and the model systems used, and not all of these residues are necessarily carcinogenic per se.
In addition, reactive metabolites of drugs play a crucial role in drug-induced liver toxicities. However, owing to their short-lived nature, they cannot be easily detected. MS-based assessments may elucidate the metabolizing path of a given drug and might indicate possible reactive metabolites, again helping with the decision of which compound series to pursue Citation[9]. Metabolically competent liver cells are crucial for the success of such metabolite studies, as well as the physiologically relevant concentration of the drug molecule Citation[10]. Based on empirical data, criteria were developed to identify ‘soft spots’ of molecules that may result in (reactive) metabolites or lead to toxic events, at least in rodent models. Other approaches that use chimeric animal models as a substitute for human pharmacokinetic studies to cut timelines early in the drug-discovery process are commercially available Citation[11]. Furthermore, efforts are being taken to build in vitro models of more than one organ connected by an artificial bloodstream in order to mimic the naturally occurring state Citation[12]. However, these handy assays do not seem to reliably be able to predict all the ADEs of clinical candidates. New innovative methods are required that are able to uncover the drug–protein interactions that lead to ADEs.
Genomics, metabolomics & proteomics in toxicology
There is evidence that the genetic background, environmental and lifestyle factors of a specific patient population determine the therapeutic window of a new medication. In these cases genomics, metabolomics and proteomics can be applied to identify the most relevant individual risk factors that will eventually determine the outcome of clinical trials. They could also help identify new biomarkers that can be used to track the progress of treatment, build up of toxicity issues and ultimate outcome. On the other hand, the gigantic amount of data resulting from ‘omics’ studies requires new data-analysis tools and is time consuming. However, this approach will ultimately lead to a better understanding of the biological system and reduce the uncertainty of a clinical outcome within a specific patient population. Integration of genomics, metabolomics and proteomics studies in a systems biology approach, the true (idiosyncratic) mode of action of drugs will be revealed. The US FDA is pursuing such combined studies comparing toxic and nontoxic drugs in relevant test systems. On the proteomic side, the quantification of the results in these studies is achieved by stable isotope-labeling techniques such as SILAC or iTRAQ Citation[13,14].
Chemical proteomics
All methods discussed above might show the critical liabilities of drugs. On investigating the data points empirically it should be possible to determine the structural features of a molecule that might cause its toxicities. However, the methods mentioned so far fail to identify a drug’s true direct target proteins that cause the liabilities. Typically, not all the interactors of a drug molecule in cells of interest are known and are not available in in vitro assays to be used to measure binding constants. Other techniques are required to find the interacting partners of the drugs.
Over the years numerous methods have evolved from the necessity to find the molecular targets of drugs and gain an insight into the interaction profile of drug molecules, in an approach now termed ‘chemical proteomics’. Four main configurations of the approach are used:
For affinity chromatography the small molecule of interest is chemically coupled to a solid-phase support, such as a polymer matrix Citation[15]. The target proteins are noncovalently bound, and nonspecific binders from a cell lysate-type sample are washed away. This method has the flipside that it leads to many false positives or negatives, and proteins that do not penetrate the polymer matrix cannot interact with the probes at all. While the false negatives will be lost, the positive hits – mostly identified by high-resolution MS – need further validation steps such as knockout animal models or siRNA experiments and this is correct for all other methods in this field. The higher the number of hits the more time-consuming the analysis will become. To get a quicker and more reliable insight into the true interaction partners of a drug molecule novel probes have been developed;
Activity-based probes (ABPs) are bifunctional small-molecule probes that form a covalent crosslink inside the pocket of the protein target through the small molecule of interest, thereby introducing a label and allowing it to be identified by SDS-PAGE analysis or MS Citation[16]. Unfortunately, not all drug molecules are available as suicidal inhibitors and limit the selection to those that can form reactive probes. Care needs to be taken so that the molecular function forming the crosslink to the protein does not interfere with the SAR of the molecule, otherwise interacting proteins will be lost or new interactions will be found;
With photo-affinity probes the target molecules are pulled out of the cell lysates and identified by MS or SDS-PAGE analysis. The photo-reactive site is located within the small molecule interacting with the protein. These modified probes need to have a strong affinity to the interacting proteins in order to obtain a high cross-linking yield, and its is difficult to synthesize these molecules;
Capture compounds are trifunctional small-molecule probes that allow a more flexible method of attaching a drug molecule to a backbone that carries different labels and functionalities, one of which is photo-inducible Citation[17]. They form a covalent crosslink with a target protein upon UV activation. The residue that forms the covalent crosslink is not located within the binding pocket. The modular system provides a new way of attaching the drug molecules in different orientations, exposing different toxico- and pharmacophoric elements and gaining insights into the complete interactome of the molecule. The third functionality (biotin) to pull out the cross-linked proteins is more distant and connected by a water-soluble linker .
Comparing the results of the same process but in the presence of an excess of drug molecules will expose the proteins that selectively interact with the drug molecule (competition control experiments, not shown).
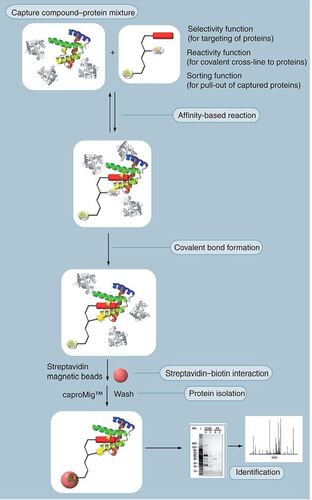
In a case study, capture compounds of tolcapone in comparison with entacapone were used. They were attached to the capture scaffold in two different orientations. With these novel probes it could be demonstrated in liver lysates that the hepatotoxic tolcapone interacts strongly with mitochondrial proteins of the respiratory chain while entacapone does not Citation[18]. Since ATP production varies with the factors determining idiosyncratic toxicities (e.g., genetic background, physical state, age and sex), it has been postulated that idiosyncratic toxicities are linked to mitochondrial dysfunction and interception with ATP synthesis. It was suggested that interference of drugs with the oxidative phosphorylation pathway is indeed a leading cause for idiosyncratic toxicity Citation[19]. Hence, it is likely that the observed interaction of tolcapone with mitochondrial membrane-bound proteins is a cause of its idiosyncratic toxicity. Pharmaceutical companies have started to test the potential of new drug candidates to interfere with the respiratory chain and other mitochondrial pathways confirming the importance of these interactions for idiosyncratic toxicities Citation[20]. Since high-resolution MS was employed in the capture experiments, all interacting proteins were directly identified in a short time. In addition, by comparison with the results of entacapone, the toxicophoric elements of tolcapone were discovered. This new technique allows an analysis of drug–protein interactions at the level of proteins endogenous to human-derived samples. This gives medicinal chemists a direct insight into the liabilities of a chemical series by flagging the molecules or parts of them as potentially dangerous. This approach requires the synthesis of novel probes Citation[21]. In the future these types of technologies might serve as complementary in vitro technologies for elucidation of drug–protein interactions. Combined with quantification techniques such as SILAC it will be possible to determine the liabilities of drug molecules and flag them for further toxicity evaluations even more accurately.
Future perspective
It has become evident that toxicities of novel drug candidates need to be assessed in humanized model systems very early in the discovery cascade in order to avoid costly failure of clinical trials at later stages. Important new in vitro techniques are evolving to guide preclinical processes, flag those molecules with possible liabilities and reduce the risk of deciding on a ‘good’ compound series. Over the next decade it will be necessary to be as successful in the prediction of (idiosyncratic) toxicities and side effects in humans as with the prediction of absorption, distribution, metabolism and excretion properties in humans 10 years ago. In the process of establishing preclinical in vitro assays, ‘omics’ technologies will play a complementary role in the evaluation of the selectivity and toxicological profile of drugs in human tissue and will be used routinely to evaluate drug molecules at early stages of the drug-discovery process. During the process it will be of significance to maintain databases that correlate the in vitro data with chemical structure and in vivo outcomes. Hazardous protein targets will be flagged as side effect-inducing anti-targets, as is already done for HERG channels. Within the coming decade new specific binding assays are going to be established to rule out interactions of drug molecules with these molecular targets, which induce ADEs in humans and animals.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
Bibliography
- Kola I , LandisJ. Can the pharmaceutical industry reduce attrition rates?Nat. Rev. Drug Discov.3(8), 711–715 (2004).
- David E , TramontinT, ZemmelR. Pharmaceutical R&D: the road to positive returns.Nat. Rev. Drug Discov.8(8), 609–610 (2009).
- Lazarou J , PomeranzBH, CoreyPN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies.JAMA279(15), 1200–1205 (1998).
- Gurwitz JH , FieldTS, AvornJet al. Incidence and preventability of adverse drug events in nursing homes. Am. J. Med. 109(2), 87–94 (2000).
- Ulrich RG . Idiosyncratic toxicity: a convergence of risk factors.Ann. Rev. Med.58, 17–34 (2007).
- Uetrecht J . Idiosyncratic drug reactions: past, present, and future.Chem. Res. Toxicol.21(1), 84–92 (2008).
- Uetrecht J . Evaluation of which reactive metabolite, if any, is responsible for a specific idiosyncratic reaction.Drug Metabo. Rev.38(4), 745–753 (2006).
- Ashby J , TennantRW. Prediction of rodent carcinogenicity for 44 chemicals: results.Mutagenesis9(1), 7–15 (1994).
- Baillie TA . Future of toxicology-metabolic activation and drug design: challenges and opportunities in chemical toxicology.Chem. Res. Toxicol.19(7), 889–893 (2006).
- Khetani SR , BhatiaSN. Microscale culture of human liver cells for drug development.Nat. Biotech.26(1), 120–126 (2008).
- Kamiya N , IwaoE, HiragaNet al. Practical evaluation of a mouse with chimeric human liver model for hepatitis C virus infection using an NS3–4A protease inhibitor. J. Gen. Virol. 91(7), 1668–1677 (2010).
- Li AP , BodeC, SakaiY. A novel in vitro system, the integrated discrete multiple organ cell culture (IdMOC) system, for the evaluation of human drug toxicity: comparative cytotoxicity of tamoxifen towards normal human cells from five major organs and MCF-7 adenocarcinoma breast cancer cells. Chem. Biol. Interac.150(1), 129–136 (2004).
- Doherty MK , HammondDE, ClagueMJ, GaskellSJ, BeynonRJ. Turnover of the human proteome: determination of protein intracellular stability by dynamic SILAC.J. Proteome Res.8(1), 104–112 (2009).
- Bantscheff M , BoescheM, EberhardD, MatthiesonT, SweetmanG, KusterB. Robust and sensitive iTRAQ quantification on an LTQ Orbitrap mass spectrometer.Mol. Cell Proteomics, 7(9), 1702–1713 (2008).
- Calleri E , TemporiniC, CaccialanzaG, MassoliniG. Target-based drug discovery: the emerging success of frontal affinity chromatography coupled to mass spectrometry.ChemMedChem4(6), 905–916 (2009).
- Salisbury CM , CravattBF. Activity-based probes for proteomic profiling of histone deacetylase complexes.Proc. Natl Acad. Sci. USA, 104(4), 1171–1176 (2007).
- Koster H , LittleDP, LuanPet al. Capture compound mass spectrometry: a technology for the investigation of small molecule protein interactions. Assay Drug Dev. Technol. 5(3), 381–390 (2007).
- Fischer JJ , MichaelisS, SchreyAKet al. Capture compound mass spectrometry sheds light on the molecular mechanisms of liver toxicity of two parkinson drugs. Toxicol. Sci. 113(1), 243–253 (2010).
- Boelsterli UA , LimPL. Mitochondrial abnormalities – a link to idiosyncratic drug hepatotoxicity?Toxicol. Appl. Pharmacol.220(1), 92–107 (2007).
- Dykens JA , MarroquinLD, WillY. Strategies to reduce late-stage drug attrition due to mitochondrial toxicity.Expert Rev. Mol. Diagn.7(2), 161–175 (2007).
- Dalhoff C , HubenM, LenzTet al. Synthesis of S-adenosyl-L-homocysteine capture compounds for selective photoinduced isolation of methyltransferases. ChemBioChem 11(2), 256–265 (2010).