
Abstract
The anti-malarial agent dihydroartemisinin (DHA) has strong anti-angiogenic activity. This study aimed to investigate the molecular mechanism underlying this effect of DHA on angiogenesis. We found that DHA shows a dose-dependent inhibition of proliferation and migration of in HUVECs. DHA specifically down-regulates the mRNA and protein expression of VEGFR2 in endothelial cells. Treatment with DHA increases IκB-α protein and blocks nuclear translocation of NF-κB p65. In addition, DHA directly regulates VEGFR2 promoter activity through p65 binding motif, and decreases the binding activity of p65 and VEGFR2 promoter, suggesting defective NF-κB signaling may underlie the observed effects of DHA on VEGFR2 expression. In the presence of the NF-κB inhibitor PDTC, DHA could not further repress VEGFR2. Co-treatment with PDTC and DHA produced minimal changes compared to the effects of either drug alone in in vitro angiogenesis assays. Similar findings were found in vivo through a mouse retinal neovascularization model examining the effects of PDTC and DHA. Our data suggested that DHA inhibits angiogenesis largely through repression of the NF-κB pathway. DHA is well tolerated, and therefore may be an ideal candidate to use clinically as an angiogenesis inhibitor for cancer treatment.
Abbreviations
| DHA | = | Dihydroartemisinin |
| VEGFR | = | Vascular Endothelial Growth Factor Receptor |
| HUVEC | = | Human Umbilical Vein Endothelial Cell |
| PDTC | = | Pyrrolidine Dithiocarbamate |
| EC | = | Endothelial Cell |
| EGF | = | Epidermal Growth Factor |
| ECIS | = | Electric Cell-Substrate Impedance Sensing |
| NF-κB | = | nuclear factor-kappa B |
Introduction
Angiogenesis is the physiological process of new capillary vessel development from pre-existing vessels. Initiation of angiogenesis and the formation of early vascular structures requires endothelial cell (EC) activation, proliferation, migration and differentiation.Citation1 Angiogenesis is essential during embryonic development, while uncontrolled angiogenesis is associated with diseases such as diabetes, rheumatoid arthritis, atherosclerosis and cancer.Citation2 The metastatic spread of cancer cells is particularly dependent on angiogenesis.Citation3 Therefore, angiogenesis inhibitors could be clinically useful for the treatment of cancer as well as other diseases dependent on new blood vessel formation.Citation3,4
A number of growth factors have been implicated in the control of angiogenesis but vascular endothelial growth factor (VEGF) is a major regulator of angiogenesis in a variety of physiological and pathological processes.Citation5,6 VEGF binds to 2 major receptor tyrosine kinases, VEGFR1 (fms-like tyrosine kinase, Flt-1) and VEGFR2 (kinase insert domain containing receptor/fetal liver kinase-1, KDR/flk-1).Citation7 Tumors derived from many human tissues secrete VEGF and its receptors are highly expressed on the endothelial cells of tumor-associated blood vessels.Citation8 Even though VEGFR2 has a weaker affinity for VEGF than VEGFR1, the cell signaling cascade activated by the VEGF/VEGFR2 interaction is responsible for the major biological effects of VEGF.Citation9 Upon VEGF binding, VEGFR2 activates a number of downstream mediators enabling endothelial cells to proliferate, migrate, invade and differentiate to form capillary-like structures.Citation10
Artemisinin is a sesquiterpene lactone endoperoxide extracted from the herbaceous plant Artemisia annua.Citation11 It is widely used as an anti-malarial with few adverse side effects.Citation12 Dihydroartemisinin (DHA), a semi-synthetic derivative of artemisinin, is more water-soluble and is an even more effective anti-malarial agent than artemisinin.Citation13 Artemisinin and its derivatives, including DHA, also display potent anticancer activities in solid tumor models.Citation14-Citation17 DHA, for example, can induce both tumor cell apoptosis and inhibit angiogenesis in lung carcinoma models.Citation18,19 A similar anti-angiogenic effect has been observed following DHA treatment in a rat embryo development model Citation20 and a chicken chorioallantoic membrane model.Citation21 In vitro, DHA can inhibit the proliferation, migration and tube formation of endothelial cells.Citation22-Citation24 Thus, DHA emerged as an anti-angiogenic component of multi-targeted cancer chemotherapies.
Several studies suggested that the anti-angiogenic effect of DHA may relate to its interaction with the VEGF signaling cascades. DHA induces a sharp decrease of VEGF expression in several human cancer cell lines.Citation21,25 In addition, DHA reduces the expression of KDR/flk-1 in the murine Lewis lung carcinoma cell line.Citation18 However, the mechanism by which DHA affects VEGF/VEGFR signaling components is not well understood.
In this study, we further investigated the effects of DHA on endothelial cell function. We found that DHA specifically reduces VEGFR2 expression, and the down-regulation of VEGFR2 is mediated by inhibition of the nuclear factor-kappa B (NF-κB) pathway. We used in vitro and in vivo models to examine the effect of the NF-κB inhibitor PDTC on DHA-mediated anti-angiogenesis. The results suggest that the anti-angiogenic effect of DHA is largely mediated through targeting the NF-κB pathway. This study expanded our understanding regarding the molecular mechanism underlying DHA's anti-angiogenic effect and supported the potential use of DHA as an agent for cancer chemotherapy or for the treatment of vascular diseases.
Results
Dose response effects of DHA on endothelial cell proliferation and migration
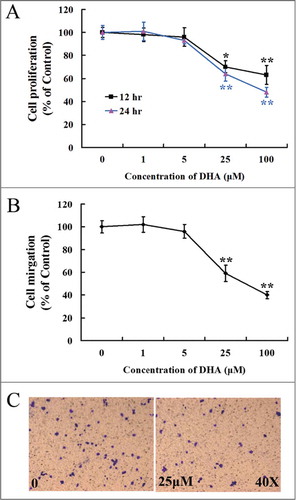
Angiogenesis is initiated from endothelial cell activation, following by cell proliferation and migration. To verify the effects of DHA on endothelial cell proliferation, MTT assay was performed for HUVECs treated with different concentrations of DHA. We found that DHA significantly reduced the growth of endothelial cells at a concentration of 25 μM or higher after 12 hr or 24 hr incubation (P < 0.05; P < 0.01; ). Boyden chamber-type transwell migration assays shown that the number of migrated cells was significantly reduced in groups treated with 25 μM DHA or higher concentration (P < 0.05; ). Therefore, DHA shows a dose-dependent inhibition of endothelial cell proliferation and migration, which also has been shown in previous studies.Citation22,26 For the next studies, we chose 25 μM for 12 hrs for the mechanism studies as it is reliably showed anti-angiogenesis effects of DHA in vitro ().
Figure 1. Dose responsive curve for endothelial cell proliferation and migration with DHA treatment. (A) MTT assay for HUVECs treated with DHA at different concentrations for 12 hrs and 24 hrs. n = 6; *, P < 0.05; **, P < 0.01; (B) Transwell migration assay for HUVECs treated with DHA at different concentrations for 12 hrs. n = 4; *, P < 0.05; **, P < 0.01. (C) Representative images of transwell migration assay treated with 0 and 25 μM DHA.
DHA down-regulates VEGFR2 expression in HUVECs
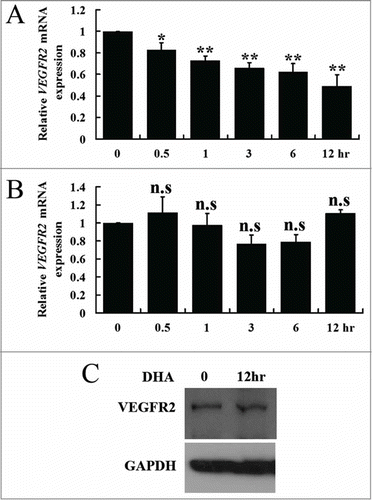
VEGFR1 and VEGFR2 are cell surface receptor tyrosine kinases (RTKs), which are expressed on endothelial cells.Citation27 Interaction between VEGF and VEGFR2 activates downstream signaling in endothelial cells that contributes to pathological angiogenesis.Citation7 In contrast, VEGFR1 is thought to inhibit angiogenesis by acting as a decoy receptor, keeping VEGF from binding VEGFR2.Citation10 We examined the expression of the VEGFR1 and VEGFR2 genes and their encoded proteins in HUVECs treated with DHA. We found that DHA did not significantly affect VEGFR1 mRNA () or VEGFR1 protein expression (). However, at a concentration of 25 μM, DHA remarkably reduced the mRNA level of VEGFR2 as early as 30 min following addition of DHA (). In lysates from HUVECs treated for 12 hr with DHA, the level of VEGFR2 protein was also significantly reduced (). We concluded that DHA specifically inhibits VEGFR2 expression, the RTK that mediates the pro-angiogenic effect of VEGF.
Figure 2. The effects of DHA onVEGFR1 and VEGFR2 expression in endothelial cells. (A) Relative VEGFR1 mRNA expression in HUVECs treated with DHA by RT-PCR. n.s., non-significant; (B) Relative VEGFR2 mRNA expression in HUVECs treated with DHA by RT-PCR. n = 4; **, P < 0.01; (C) Representative immunoblot of VEGFR1 in DHA treated HUVECs; (D) Representative immunoblot of VEGFR2 in DHA treated HUVECs.
DHA inhibits NF-κB signaling in HUVECs
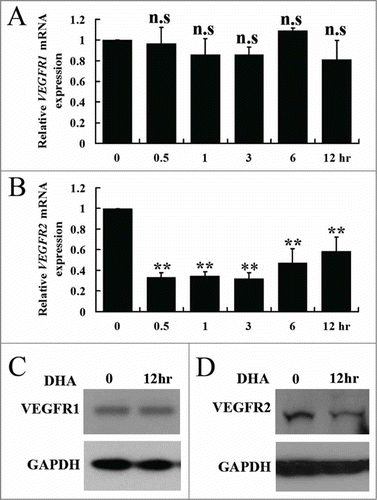
NF-κB signaling mediates numerous cellular processes and positively regulates VEGFR2 expression.Citation28 Activation of NF-κB requires the degradation of inhibitor of kappa B (IκB-α), which forms a cytoplasmic and inactive complex with the p65-p50 heterodimer. The complex is inactive because IκB-α is able to block the nuclear localization signals (NLS) of the NF-κB subunits, keeping the complex outside nucleus.Citation29,30 We separated the cytoplasm and nucleus of HUVECs, and assessed whether DHA influences IκB-α and NF-κB p65 by Western blot. The protein level of IκB-α in cytoplasm was remarkably increased, while the NF-κB p65 in nucleus was significantly decreased 6 hrs after the addition of DHA (). This observation suggested that DHA could function through inhibition of the NF-κB signaling pathway in endothelial cells.
Figure 3. DHA inhibits NF-κB pathway in endothelial cells. (A) Representative immunoblots of IκB-α (cytoplasmic) and NF-κB p65 (nuclear) extracted from HUVECs treated with DHA for 6 hrs. GAPDH and Histone H3 were used as loading control for cytoplasmic and nuclear protein, respectively. (B) Immunoflourescence staining of NF-κB p65 in HUVECs. Arrows refer to positive p65 staining in the nucleus.
Epidermal growth factor (EGF) stimulates NF-κB translocation through an IκB-α independent mechanism, but the translocation can be blocked by an increase of IκB-α protein.Citation31,32 We next examined whether DHA could inhibit EGF-induced NF-κB translocation in endothelial cells. Using immunofluorescence imaging of serum starved, EGF-treated HUVECs, we were able to demonstrate the nuclear translocation of the NF-κB p65 subunit (). In contrast, DHA treatment results in cytosolic retention and a lack of nuclear p65 staining. These data suggested that DHA inhibits NF-κB pathway through blockage of NF-κB translocation, presumably by increasing the level of IκB-α protein in endothelial cells.
DHA reduced VEGFR2 promoter activity via NFkB binding motif
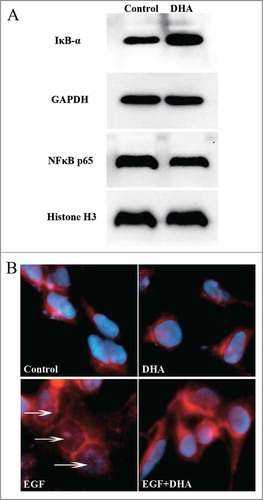
The human VEGFR2 promoter contains a number of consensus binding sites for transcriptional regulation.Citation33 Previous studies have suggested that the -80 NF-κB-binding site is essential in maintaining constitutive expression of VEGFR2.Citation34 We generated the mutation of the NF-κB-binding site on human VEGFR2 promoter and examined the promoter activity by transient transfection assay. showed that the mutation resulted in 67.23% decrease of the promoter activity (P < 0.01). The promoter activities were further examined after treatment with different concentrations of DHA for 12 hrs. DHA significantly reduced the activity of VEGFR2 promoter at a concentration of 25 μM or higher (, P < 0.05). However, DHA failed to affect the activity of VEGFR2 promoter with the mutation of the NF-κB bind site at a concentration of 100 μM (). Therefore, DHA downregulates VEGFR2 promoter activity through the mediation of NF-κB binding motif.
Figure 4. DHA regulates VEGFR2 promoter activity via NF-κB binding motif. (A) Comparison of the activity of VEGFR2 promoter with and without the mutation of NF-κB binding site. n = 6, ** P < 0.01; (B) The effects of DHA on VEGFR2 promoter activity. n = 6, * P < 0.05; (C) The effects of DHA on the activity of VEGFR2 promoter with the mutation of NF-κB binding site. n = 6. (D) ChIP assay for p65 binding to VEGFR2 promoter in HUVEC with and without DHA treatment. (E) Binding ratio relative to total input chromatin in the ChIP reaction.
DHA reduced the binding of p65 protein to VEGFR2 promoter
To examine effects of DHA on the binding activities of NF-κB p65 protein to the DNA elements on VEGFR2 promoter, ChIP assay was performed using HUVECs treated in the absence or presence of 25 μM DHA for 12 hrs. Native chromatin was immunoprecipitated with antibodies against p65 or control IgG, and the immunoprecipitated fragments were subjected to PCR using specific primers flanking a region including the NF-κB binding site on the immediate upstream promoter of VEGFR2. As shown in and E, the VEGFR2 promoter was bound by p65, and the level of binding was significantly reduced by DHA treatment.
The NF-κB inhibitor PDTC compromises DHA induced VEGFR2 repression
PDTC is a potent inhibitor of the NF-κB pathway.Citation35 PDTC prevents IκB-α degradation, precluding the dissociation of NF-κB from IκB-α and thus NF-κB's translocation to the nucleus.Citation36 At a concentration of 100 μM, PDTC significantly reduced the expression of VEGFR2 for up to 12 hrs (). To determine if DHA and PDTC share a similar mechanism by which they can repress VEGFR2, we pre-treated HUVECs with 100 μM PDTC for 1 hr before measuring the effect of DHA on VEGFR2 expression. Following PDTC pre-treatment, addition of DHA could not further repress the expression of VEGFR2 (). In addition, we examined the protein expression of VEGFR2 by western blot. At 12 hr, the protein level of VEGFR2 in PDTC/DHA treated HUVECs was similar to that of cells treated with PDTC alone (). Since there was no synergistic or additive effect of DHA and PDTC in the repression of VEGFR2, the data suggest that both agents repress VEGFR2 via an effect on NF-κB signaling.
Figure 5. The effects of PDTC on VEGFR2 expression in endothelial cells treated with DHA. (A) Relative VEGFR2 mRNA expression in HUVECs treated with PDTC. n = 4; *, P < 0.05; **, P < 0.01; (B) Relative VEGFR2 mRNA expression in HUVECs treated with DHA after PDTC pretreatment. n = 4; n.s., non-significant. (C) Representative immunoblot of VEGFR2 in DHA treated HUVECs after PDTC pre-treatment.
Impaired NF-κB signaling mediates DHA-induced repression of endothelial cell function
The effects of PDTC on DHA-induced repression of HUVEC proliferation was examined by MTT assay. After a 12 hr incubation, DHA or PDTC alone significantly reduced HUVEC growth (P < 0.01; P < 0.01; ). The combination of DHA and PDTC reduced HUVEC cell number by 52.23%. This was only an additional 5.24% decrease from the reduction caused by PDTC alone (46.99%, ). It would implicate that once the NF-κB pathway is blocked, the effect of DHA is neither synergistic nor additive to the effect of PDTC on HUVEC proliferation. To confirm this finding we carried out real-time proliferation experiments using cultures of HUVECs with ECIS, which measured the electrical resistance over a monolayer of endothelial cells. In these experiments, ECIS readings are proportional to cell density in the culture well. With DHA, PDTC and combination treatment, a reduction in electrical resistance was measured at all time points (4, 8, 12 and 16 hrs) (). These results were similar to the MTT assay. The combination of DHA and PDTC showed only a small difference compared to PDTC treatment alone. This suggested endothelial cell proliferation is impaired by DHA via the NF-κB pathway.
Figure 6. The effects of PDTC on DHA induced repression of angiogenesis. (A) MTT assay for HUVECs treated with PDTC and DHA for 12 hrs. n = 6; *, P < 0.05; **, P < 0.01; (B) ECIS proliferation assay. The data are shown relative to control cells and the bar graphs represent the average of 3 repeats at the indicated time points. (C) Transwell migration assay for HUVECs treated with PDTC and DHA for 12 hrs. n = 4; **, P < 0.01; n.s., non-significant; (D) ECIS wound healing assay. The data are shown relative to control cells and the bar graphs represent the average of 3 repeats at the indicated time points. (E and F) Angiographic analysis of the effect of PDTC and DHA on retinal neovascularization. (E) Representative image of FITC-isolectin staining of PI7 retinal blood vessels from eyes of hypoxic mice with PBS,DHA,PDTC and PDTC plus DHA; (F) Relative retinal vessel density of the hypoxic mice treated with DHA with and without PDTC co-treatment. n = 6; **, P < 0.01; n.s., non-significant.
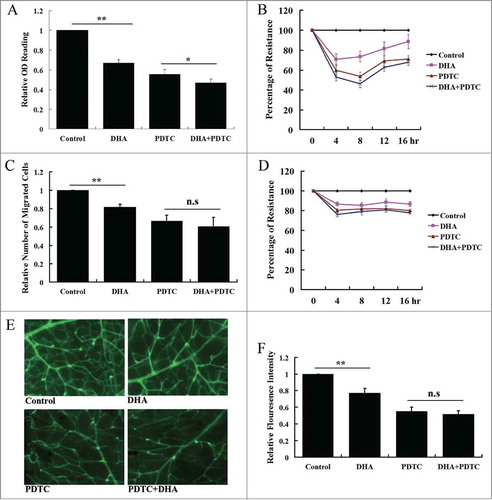
Transwell migration assays were used to assess the effects of DHA, PDTC and their combination on endothelial cell migration. The number of cells migrating across the layer of matrigel was significantly reduced in the DHA (18.12%, P < 0.01) and PDTC (33.58%, P < 0.01) treated groups compared to cells treated with vehicle alone (). The combination of DHA and PDTC reduced cell migration by 39.34%; however, this was not a significant difference between the combination treatment and cells treated with PDTC alone (p = 0.13). We also performed real-time wound healing assays by ECIS. At all time points, HUVECs treated with PDTC and PDTC together with DHA showed a similar resistance. Both inhibited re-filling of the well-defined wounds made by the ECIS electrode to a similar extent (). These in vitro assays indicated that inhibition of NF-κB pathway compromised DHA induced reduction of endothelial cell proliferation and migration.
The effects of DHA and PDTC on mouse retinal neovascularization
We wondered if the inhibitory effect of DHA and PDTC observed using in vitro assays of endothelial cell function could be recapitulated in vivo. Therefore, we assayed their anti-angiogenic effects using a retinal neovascularization model. In this model, neonatal mice are placed under hyperoxic conditions, which lead to the extensive obliteration of retinal capillaries. When the mice are returned to basal levels of oxygen, the inner retina remains relatively hypoxic compared to the outer normoxic conditions. This stimulates the production of specific cytokines that induce retinal neovascularization, and NF-κB pathway is activated under these conditions.Citation37 In our study, intravitreal injections of DHA and PDTC were successfully performed on P12 mice immediately following the exposure to hyperoxia. After return to normoxia for 5 days, retinas from DHA-treated eyes showed a reduction in vessel density (36.56%, P < 0.01) compared with vehicle control treated mice (, top panels; ). Retinas from PDTC-treated eyes also showed a significant reduction in vessel density (48.23%; P < 0.01). When both DHA and PDTC were given as the treatment, retinas from mice receiving the DHA/PDTC combination showed a statistically non-significant reduction in vessel density compared to mice receiving PDTC alone (, lower panels; ; p = 0.16). Thus, the effects of DHA were largely mediated by inhibition of the NF-κB pathway. The observed effects of the combination were neither synergistic nor additive in vivo.
Discussion
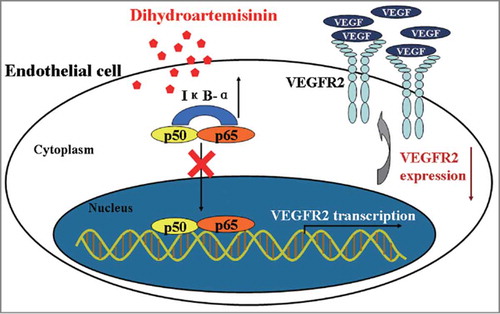
Artemisinin and its derivatives have shown promise in pre-clinical models as anti-cancer drugs. A considerable amount of research has focused on the most active derivatives of artemisinin, including DHA.Citation38 DHA shows remarkable anti-tumor activity against pancreatic adenocarcinoma, leukemia, osteosarcoma, and lung carcinoma cells.Citation14 DHA also inhibits tumor angiogenesis, but the molecular mechanism remains enigmatic. In this investigation we found that DHA down-regulates VEGFR2 mRNA and VEGFR2 protein expression in endothelial cells. We also demonstrated that DHA treatment can increase IκB-α and inhibit NF-κB p65 nuclear translocation in endothelial cells. In addition, we confirmed that DHA regulates VEGFR2 promoter activity through NF-κB binding motif, and decreases the binding activity of p65 and VEGFR2 promoter. Using various assays dependent on NF-κB function, we demonstrated that the NF-κB inhibitor PDTC, which itself inhibits NF-κB and leads to VEGFR2 downregulation, obviates any inhibitory effect of DHA in both in vitro and in vivo models of angiogenesis. Our findings suggest that DHA inhibits VEGFR2 expression and angiogenesis largely through inhibition of the NF-κB pathway ().
Figure 7. Schematic illustration of the mechanisms of the anti-angiogenesis effects of DHA.
Delivery of anti-angiogenesis is one therapeutic approach that holds promise for the treatment of highly malignant tumors.Citation39 The VEGF/VEGFR2 signaling axis, as a major regulator of angiogenesis, is commonly targeted by anti-angiogenic therapies. In this study, we found that DHA treatment leads to down-regulation of VEGFR2, but not VEGFR1 expression in endothelial cells. This suggests that VEGFR2 is a downstream target of DHA that mediates its anti-angiogenic effect. Consistent with our findings, Zhou et al., found that DHA suppresses the expression of VEGFR2 in Lewis lung carcinoma cells.Citation18 Although VEGFR2 is expressed at low levels in some tumor cells, such as the Lewis lung carcinoma cells, VEGFR2 expression is highly enriched in endothelial cells. Indeed it is well accepted as an endothelial cell marker.Citation40
Our findings suggest that blockade of NF-κB signaling in endothelial cells mediates the anti-angiogenic properties of DHA. Consistently, NF-κB signaling is associated with the pathological angiogenesis observed in the vasculature of colorectal and lung carcinomaCitation41,42 and strategies to inhibit NF-κB are effective in decreasing tumor angiogenesis. For example, the anti-angiogenic properties of thalidomide have been attributed to its ability to inhibit the NF-κB pathway.Citation43 VEGFR2 has been identified as a downstream target gene regulated by the NF-κB pathway. NF-κB directly binds the VEGFR2 upstream promoter and activates its transcription.Citation28 Previously it has been shown that DHA can prevent the degradation of IκB-α protein in cancer cell lines.Citation26,36 The current study demonstrates that DHA also increases IκB-α protein in endothelial cells, and blocks translocation of p65 into the nucleus. We also discovered that DHA regulates VEGFR2 promoter activity through p65 binding motif, and decreases the binding activity of p65 and VEGFR2 promoter. In addition, we found that the NF-κB inhibitor PDTC obviates DHA-induced VEGFR2 downregulation. This is the first study connecting these 3 effects of DHA, inhibition of NF-κB, down-regulation of VEGFR2 and anti-angiogenesis. Our findings provide mechanistic insights into the anti-angiogenic properties of DHA.
Inhibiting NF-κB function has been proposed as a possible adjuvant to traditional chemotherapeutic strategies.Citation44 In addition to VEGFR2, NF-κB also targets numerous pro-proliferativeCitation45 and angiogenic stimulators.Citation46 In our study, we found DHA caused an additional anti-proliferative effect in the endothelial cells pretreated with PDTC. This suggests that DHA may also affect other genes and signaling pathways in endothelial cells. Previously it has been shown that artemisinin can directly activate apoptotic pathways in endothelial cells.Citation47 In a rat corneal neovascularization model, DHA inhibits suture-induced angiogenesis through the ERK1/2 and p38 pathways.Citation48 Taken together, these findings and ours suggest that inhibition of the NF-κB pathway plays major role in mediating the anti-angiogenic effects of DHA, while other pathways may also participate in this process.
Retinal neovascularization is a major cause of the blindness associated with diabetic retinopathy, retinal vein occlusion and age-related macular degeneration.Citation49 The model of ischemia-induced retinal neovascularization that we utilized is a well-accepted in vivo angiogenesis model.Citation50 To our knowledge this is the first study demonstrating that DHA significantly inhibits retinal neovascularization. This suggests the potential in using DHA to treat vascular eye diseases. Inhibition of retinal neovascularization by PDTC has been previously reported.Citation37 The current study indicates that the combination of DHA and PDTC treatment showed only a limited decrease of retinal neovascularization comparing to the treatment with PDTC alone. This is consistent with our in vitro functional assays shown that inhibition of the NF-κB pathway is the key mechanism by which DHA affects angiogenesis.
DHA is an emerging candidate as a clinical angiogenesis inhibitor due to its anti-angiogenic properties and its known low toxicity. Our data shed light on the molecular mechanism that underlies the anti-angiogenic activities of DHA, which will be important for further exploration of its clinic application. Our results further strengthened the potential use of the artemisinin family of compounds as therapeutic agents against cancer and other vascular-related diseases.
Materials and Methods
Cell culture
HUVECs were purchased from Lonza and cultured in basal endothelial cell medium (EBM2) supplemented using the EGM-2-MV bullet kit (Lonza) and antibiotics (100 IU/ml penicillin and 100 μg/ml streptomycin). The cells were cultured in humidified air at 37°C with 5% CO2. DHA (D7439, Sigma-Aldrich) and PDTC (P8765, Sigma-Aldrich) were dissolved in DMSO.
Western blotting
HUVECs were washed with PBS and lysed in ice-cold RIPA buffer (20 mM Tris pH 7.5, 150 mM NaCl, 50 mM NaF, 1% NP40, 0.1% DOC, 0.1% SDS, 1 mM EDTA, 1 mM PMSF, 1 μg/ml leupeptin). For examination of IκBα and NF-κB p65, the cytoplasm and nucleus were separated with NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific). Protein concentration was determined using the BCA assay (Bio-Rad). Equal amounts of protein were separated by SDS-PAGE (8% polyacrylamide gel) and transferred on to a PVDF membrane. PVDF membranes were blocked with 2.5% BSA and incubated overnight with primary antibody in PBS-T at 4°C. Primary antibodies included rabbit anti-VEGFR2 (92006, Novus Biologicals), rabbit anti-VEGFR1 (H00002321 Novus Biologicals), mouse anti-IκBα (L35A5, Cell Signaling), mouse anti-NF-κB p65 (6956, Cell Signaling), rabbit anti-Histone H3 antibody (9715, Cell Signaling) and anti-GAPDH (14C10, Cell Signaling) antibodies. Immunoreactivity was visualized with HRP-conjugated secondary antibodies and chemiluminescence. GAPDH levels were used as controls for protein loading.
Immunofluorescence
Serum-starved cultures of HUVECs were grown on coverslips. These cells were incubated with 25 μM DHA or 100 nM EGF. After 2 hrs of stimulation, cells were fixed for 1 min in 95% ethanol and subsequently incubated for 10 min in 4% paraformaldehyde at room temperature. Cells were stained with a rabbit polyclonal NF-κB p65 (Abcam) antibody for 1 hr and detected with an Alexa546-labeled anti-rabbit secondary antibody (Molecular Probes). Nuclear staining was achieved with DAPI (Molecular Probes) and samples were imaged by with a fluorescence microscope (Olympus).
Quantitative real-time PCR (qRT-PCR)
RNA isolation and cDNA synthesis were performed using the RNeasy Mini kit (Qiagen) and High Capacity RNA-to-cDNA Master Mix (Applied Biosystems) respectively. qRT-PCR was performed using a ViiA7 Real-Time PCR System (Applied Biosystems). Reactions conditions were 95°C for 5 min, 40 cycles of 95°C for 10 sec, 60°C for 20 sec and 72°C for 5 sec. All PCR reactions were repeated in triplicate. Relative expression was calculated using GAPDH as an endogenous internal control. The primer sequences were as follows:
VEGFR1 forward primer, 5′-CACCAAGAGCGACGTGTG-3′
VEGFR1 reverse primer, 5′-TTTTGGGTCTCTGTGCCAG-3′
VEGFR2 forward primer, 5′-CACCACTCAAACGCTGACATGTA-3′
VEGFR2 reverse primer 5′-GCTCGTTGGCGCACTCTT-3′
GAPDH forward primer, 5′-TGATGACATCAAGAAGGTGGTGAAG-3′
GAPDH reverse primer, 5-TCCTTGGAGGCCATGTGGGCCAT-3′
Proliferation assays
Cell proliferation was evaluated with MTT assay kit (Cayman). Briefly, cells were seeded at a density of 5 × 103 cells/well in a 96-well plate. The next day, DHA (0, 1, 5, 25, 100 μM), PDTC (100 μM) or a combination of both compounds was added to the culture media. After 12 or 24 hrs, cells were washed with PBS and MTT solution (10 μl of 5 mg/mL) was added to each well for 2 hrs. Formazan crystals were solubilized and colorimetric intensity was analyzed using a 96-well plate reader (Molecular Devices) at a wavelength of 570 nm. Each experiment was repeated in triplicate.
Cell migration assays
Transwell migration assays were performed using the QCM 3 μm Endothelial Cell Migration Assay Fibronectin kit (Millipore). Briefly, 1 × 105 HUVECs in 300 μl of EBM2 containing DHA and/or PDTC were seeded into the upper chambers of the transwells. The bottom wells of the chambers were filled with 500 μl of EBM2 supplemented using the EGM-2-MV bullet kit. After 12 h of incubation at 37°C, the migrated cells were fixed and stained with 0.1% crystal violet. Migration was quantified by counting the number of stained cells at 100× magnification with a phase-contrast microscope (Olympus).
Plasmids and transient transfection
The −115 to +296 fragment of VEGFR2 promoter was generated by DNA synthesis and inserted into pGL3 vector by KpnI and XhoI sites. The −90 NF-κB mutation (GGAGAGCCCC → AAAGAGCCTT) on the VEGFR2 promoter was generated by PCR. HUVECs were transfected using Lipofectamine3000 (Invitrogen) as instructed by the manufacturer. A plasmid containing the Renilla luciferase reporter gene under the control of a cytomegalovirus enhancer/promoter (pRL-CMV) (Promega) was used as internal control. 24 h after transfection, the cells were washed with PBS and cultured for 12 h in EBM2 plus 0.5% FBS. The cells were then incubated in different concentrations of DHA for 12 h. Then the cells were lysed and assayed for luciferase activity using the dual-luciferase reporter assay system (Promega).
Chromatin immunoprecipitation (ChIP) assay
HUVECs were exposed to 2% paraformaldehyde for 10 min and then treated with 0.2 M glycine for 5 min. The cells were washed twice with PBS containing a protease inhibitor cocktail (Roche), then scraped and collected by centrifugation. The cell pellets were sonicated with a Branson 250 Sonifier to shear the chromatin into 300–600 bp fragments. Immunoprecipitation was performed using a ChIP assay kit (Upstate Biotechnology Inc.) according to the manufacturer's instructions. The chromatin fragments were immunoprecipitated with the antibody against NF-κB p65 (Abcam). The DNA fragments were detected by real-time PCR with primers as follows: forward: 5′-CCAGCGCAGTCCAGTTGTGT-3′; reverse: 5′-CGAAACTCTAGAGCGCGGAG-3′.
Electric cell-substrate impedance sensing (ECIS) analysis
The intercellular impedance across a monolayer of endothelial cells was measured using the ECIS technique (ECIS model 1600; Applied BioPhysics). Briefly, 8-well ECIS arrays (8W10E+) were coated with L-cysteine. HUVECs were plated in the wells and allowed to form monolayers. DHA and/or PDTC were added to the wells and the resistance across the EC layer was acquired. For wound healing assays, the AC current was delivered to the small active electrodes located on the central part of each well. Cells located near the electrodes were killed and detached from the monolayer. The wounded areas of each well were gradually healed by the migration of viable cells surrounding the wounded area and the migratory response was measured in real-time by recording the electrical impedance. Data plots are representative of triplicate experiments, with each graph showing impedance readings from a separate well, at 40 distinct electrodes per well.
Animal model of retinal neovascularization and retinal vessel density examinations
All experiments were institutionally approved and conformed to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. We utilized a model of hypoxia-induced neovascularization that has been described previously.Citation37 Litters of postnatal day 7 (P7) C57BL/6N mice, together with their mothers, were exposed to 75% oxygen for 5 d and then returned to basal levels of oxygenation on P12. Intravitreal injections were performed daily from P12 to P14 by delivering 0.5 μl of 0.1 mM DHA diluted in PBS to the left eye and PBS alone to the right eye, using a 32-gauge needle at 200 μm posterior to the limbus. A second group of mice received 0.5 μl of 0.1 mM DHA and 0.5 mM of PDTA to the left eye, and 0.5 mM of PDTA to the right eye. The mice were sacrificed on P17 and retinas were removed for vessel density examination. Briefly, eyes were fixed with 4% paraformaldehyde for 30 min and cut 1.5 mm behind the limbus. The anterior segments including lens, cornea, and iris were removed. The retina was separated from the sclera and flattened by making 4 radial cuts. The vitreous were gently removed from the retinal surface using forceps. Retinas were subsequently incubated with 0.5% fluorescein isothiocyanate (FITC)-isolectin B4 (Vector Laboratories) overnight to allow for visualization of vascular structure using a fluorescence microscope (Olympus). Retinal images were processed and quantified using ImageJ software.
Statistical analysis
Data were expressed as mean ± standard error (SE). Comparison of means was by the unpaired, 2-tailed Student's t-test.
Disclosure of Potential Conflicts of Interest
The authors declare that there are no potential conflicts of interest.
Funding
This study was supported by a grant from Medical Science and Technology Development Plan of Shandong Province (2013WS0137) and Shandong Taishan Scholarship (Ju Liu, Suhua Yan).
References
- Risau W. Mechanisms of angiogenesis. Nature 1997; 386:671–4; PMID:9109485; http://dx.doi.org/10.1038/386671a0
- Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995; 1:27-31; PMID:7584949; http://dx.doi.org/10.1038/nm0195-27
- Carmeliet P. Angiogenesis in life, disease and medicine. Nature 2005; 438:932-6; PMID:16355210; http://dx.doi.org/10.1038/nature04478.
- Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature 2005; 438:967-74; PMID:16355214; http://dx.doi.org/10.1038/nature04483.
- Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature 2005; 438:937-45; PMID:16355211; http://dx.doi.org/10.1038/nature04479.
- Senger DR, Van de Water L, Brown LF, Nagy JA, Yeo KT, Yeo TK, Berse B, Jackman RW, Dvorak AM, Dvorak HF. Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer Metastasis Rev 1993; 12:303-24; PMID:8281615; http://dx.doi.org/10.1007/BF00665960.
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003; 9:669-76; PMID:12778165; http://dx.doi.org/10.1038/nm0603-669.
- Kolch W, Martiny-Baron G, Kieser A, Marme D. Regulation of the expression of the VEGFVPS and its receptors: role in tumor angiogenesis. Breast Cancer Res Treat 1995; 36:139-55; PMID:8534863; http://dx.doi.org/10.1007/BF00666036.
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011; 473:298-307; PMID:21593862; http://dx.doi.org/10.1038/nature10144.
- Sato Y, Kanno S, Oda N, Abe M, Ito M, Shitara K, Shibuya M. Properties of two VEGF receptors, Flt-1 and KDR, in signal transduction. Ann N Y Acad Sci 2000; 902:201-5; discussion 5-7; PMID:10865839; http://dx.doi.org/10.1111/j.1749-6632.2000.tb06314.x.
- Klayman DL. Qinghaosu (artemisinin): an antimalarial drug from China. Science 1985; 228:1049-55; PMID:3887571; http://dx.doi.org/10.1126/science.3887571.
- Gordi T, Lepist EI. Artemisinin derivatives: toxic for laboratory animals, safe for humans? Toxicol Lett 2004; 147:99-107; PMID:14757313; http://dx.doi.org/10.1016/j.toxlet.2003.12.009.
- Tu Y. The development of new antimalarial drugs: qinghaosu and dihydro-qinghaosu. Chin Med J (Engl) 1999; 112:976-7; PMID:11721477.
- Crespo-Ortiz MP, Wei MQ. Antitumor activity of artemisinin and its derivatives: from a well-known antimalarial agent to a potential anticancer drug. J Biomed Biotechnol 2012; 2012:247597; PMID:22174561; http://dx.doi.org/10.1155/2012/247597.
- Efferth T, Dunstan H, Sauerbrey A, Miyachi H, Chitambar CR. The anti-malarial artesunate is also active against cancer. Int J Oncol 2001; 18:767-73; PMID:11251172.
- Dell’Eva R, Pfeffer U, Vene R, Anfosso L, Forlani A, Albini A, Efferth T. Inhibition of angiogenesis in vivo and growth of Kaposi's sarcoma xenograft tumors by the anti-malarial artesunate. Biochem Pharmacol 2004; 68:2359-66; http://dx.doi.org/10.1016/j.bcp.2004.08.021.
- Zhang CZ, Zhang H, Yun J, Chen GG, Lai PB. Dihydroartemisinin exhibits antitumor activity toward hepatocellular carcinoma in vitro and in vivo. Biochem Pharmacol 2012; 83:1278-89; PMID:22342732; http://dx.doi.org/10.1016/j.bcp.2012.02.002.
- Zhou HJ, Zhang JL, Li A, Wang Z, Lou XE. Dihydroartemisinin improves the efficiency of chemotherapeutics in lung carcinomas in vivo and inhibits murine Lewis lung carcinoma cell line growth in vitro. Cancer Chemother Pharmacol 2010; 66:21-9; PMID:19756601; http://dx.doi.org/10.1007/s00280-009-1129-z.
- Lu JJ, Meng LH, Shankavaram UT, Zhu CH, Tong LJ, Chen G, Lin LP, Weinstein JN, Ding J. Dihydroartemisinin accelerates c-MYC oncoprotein degradation and induces apoptosis in c-MYC-overexpressing tumor cells. Biochem Pharmacol 2010; 80:22-30; PMID:20206143; http://dx.doi.org/10.1016/j.bcp.2010.02.016.
- Longo M, Zanoncelli S, Torre PD, Riflettuto M, Cocco F, Pesenti M, Giusti A, Colombo P, Brughera M, Mazue G, et al. In vivo and in vitro investigations of the effects of the antimalarial drug dihydroartemisinin (DHA) on rat embryos. Reprod Toxicol 2006; 22:797-810; PMID:16959470; http://dx.doi.org/10.1016/j.reprotox.2006.08.001.
- Chen HH, Zhou HJ, Wang WQ, Wu GD. Antimalarial dihydroartemisinin also inhibits angiogenesis. Cancer Chemother Pharmacol 2004; 53:423-32; PMID:15132130; http://dx.doi.org/10.1007/s00280-003-0751-4.
- Chen HH, Zhou HJ, Fang X. Inhibition of human cancer cell line growth and human umbilical vein endothelial cell angiogenesis by artemisinin derivatives in vitro. Pharmacol Res 2003; 48:231-6; PMID:12860439; http://dx.doi.org/10.1016/S1043-6618(03)00107-5.
- Wu GD, Zhou HJ, Wu XH. Apoptosis of human umbilical vein endothelial cells induced by artesunate. Vascular Pharmacol 2004; 41:205-12; PMID:15653096; http://dx.doi.org/10.1016/j.vph.2004.11.001.
- D’Alessandro S, Basilico N, Corbett Y, Scaccabarozzi D, Omodeo-Sale F, Saresella M, Marventano I, Vaillant M, Olliaro P, Taramelli D. Hypoxia modulates the effect of dihydroartemisinin on endothelial cells. Biochem Pharmacol 2011; 82:476-84; http://dx.doi.org/10.1016/j.bcp.2011.06.002.
- Zhou HJ, Wang WQ, Wu GD, Lee J, Li A. Artesunate inhibits angiogenesis and downregulates vascular endothelial growth factor expression in chronic myeloid leukemia K562 cells. Vascular Pharmacol 2007; 47:131-8; PMID:17581794; http://dx.doi.org/10.1016/j.vph.2007.05.002.
- Wang SJ, Sun B, Cheng ZX, Zhou HX, Gao Y, Kong R, Chen H, Jiang HC, Pan SH, Xue DB, et al. Dihydroartemisinin inhibits angiogenesis in pancreatic cancer by targeting the NF-kappaB pathway. Cancer Chemother Pharmacol 2011; 68:1421-30; PMID:21479633; http://dx.doi.org/10.1007/s00280-011-1643-7.
- Terman BI, Dougher-Vermazen M. Biological properties of VEGFVPF receptors. Cancer Metastasis Rev 1996; 15:159-63; PMID:8842487; http://dx.doi.org/10.1007/BF00437468.
- Ronicke V, Risau W, Breier G. Characterization of the endothelium-specific murine vascular endothelial growth factor receptor-2 (Flk-1) promoter. Circ Res 1996; 79:277-85; PMID:8756005; http://dx.doi.org/10.1161/01.RES.79.2.277.
- Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkappaBalphaNF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell 1998; 95:759-70; PMID:9865694; http://dx.doi.org/10.1016/S0092-8674(00)81699-2.
- Jacobs MD, Harrison SC. Structure of an IkappaBalphaNF-kappaB complex. Cell 1998; 95:749-58; PMID:9865693; http://dx.doi.org/10.1016/S0092-8674(00)81698-0.
- Sethi G, Ahn KS, Chaturvedi MM, Aggarwal BB. Epidermal growth factor (EGF) activates nuclear factor-kappaB through IkappaBalpha kinase-independent but EGF receptor-kinase dependent tyrosine 42 phosphorylation of IkappaBalpha. Oncogene 2007; 26:7324-32; PMID:17533369; http://dx.doi.org/10.1038/sj.onc.1210544.
- Haussler U, von Wichert G, Schmid RM, Keller F, Schneider G. Epidermal growth factor activates nuclear factor-kappaB in human proximal tubule cells. Am J Physiol Renal Physiol 2005; 289:F808-15; PMID:15798085; http://dx.doi.org/10.1152/ajprenal.00434.2003.
- Patterson C, Perrella MA, Hsieh CM, Yoshizumi M, Lee ME, Haber E. Cloning and functional analysis of the promoter for KDRflk-1, a receptor for vascular endothelial growth factor. J Biol Chem 1995; 270:23111-8; PMID:7559454; http://dx.doi.org/10.1074/jbc.270.39.23111.
- Minami T, Rosenberg RD, Aird WC. Transforming growth factor-beta 1-mediated inhibition of the flk-1KDR gene is mediated by a 5’-untranslated region palindromic GATA site. J Biol Chem 2001; 276:5395-402; PMID:11098056; http://dx.doi.org/10.1074/jbc.M008798200.
- Ziegler-Heitbrock HW, Sternsdorf T, Liese J, Belohradsky B, Weber C, Wedel A, Schreck R, Bauerle P, Strobel M. Pyrrolidine dithiocarbamate inhibits NF-kappa B mobilization and TNF production in human monocytes. J Immunol 1993; 151:6986-93; PMID:8258705.
- Liu SF, Ye X, Malik AB. Pyrrolidine dithiocarbamate prevents I-kappaB degradation and reduces microvascular injury induced by lipopolysaccharide in multiple organs. Mol Pharmacol 1999; 55:658-67; PMID:10101023.
- Yoshida A, Yoshida S, Ishibashi T, Kuwano M, Inomata H. Suppression of retinal neovascularization by the NF-kappaB inhibitor pyrrolidine dithiocarbamate in mice. Invest Ophthalmol Vis Sci 1999; 40:1624-9; PMID:10359349.
- Ho WE, Peh HY, Chan TK, Wong WS. Artemisinins: Pharmacological actions beyond anti-malarial. Pharmacol Ther 2013; PMID:24316259.
- Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 2006; 12:895-904; PMID:16892035; http://dx.doi.org/10.1038/nm1469.
- Yamaguchi TP, Dumont DJ, Conlon RA, Breitman ML, Rossant J. flk-1, an flt-related receptor tyrosine kinase is an early marker for endothelial cell precursors. Development 1993; 118:489-98; PMID:8223275.
- DeBusk LM, Massion PP, Lin PC. IkappaB kinase-alpha regulates endothelial cell motility and tumor angiogenesis. Cancer Res 2008; 68:10223-8; PMID:19074890; http://dx.doi.org/10.1158/0008-5472.CAN-08-1833.
- Sakamoto K, Maeda S, Hikiba Y, Nakagawa H, Hayakawa Y, Shibata W, Yanai A, Ogura K, Omata M. Constitutive NF-kappaB activation in colorectal carcinoma plays a key role in angiogenesis, promoting tumor growth. Clin Cancer Res: An Off J Am AssocCancer Res 2009; 15:2248-58; PMID:19276252; http://dx.doi.org/10.1158/1078-0432.CCR-08-1383.
- Keifer JA, Guttridge DC, Ashburner BP, Baldwin AS, Jr. Inhibition of NF-kappa B activity by thalidomide through suppression of IkappaB kinase activity. J Biol Chem 2001; 276:22382-7; PMID:11297551; http://dx.doi.org/10.1074/jbc.M100938200.
- Calzado MA, Bacher S, Schmitz ML. NF-kappaB inhibitors for the treatment of inflammatory diseases and cancer. Curr Med Chem 2007; 14:367-76; PMID:17305539; http://dx.doi.org/10.2174/092986707779941113.
- Brouard S, Berberat PO, Tobiasch E, Seldon MP, Bach FH, Soares MP. Heme oxygenase-1-derived carbon monoxide requires the activation of transcription factor NF-kappa B to protect endothelial cells from tumor necrosis factor-alpha-mediated apoptosis. J Biol Chem 2002; 277:17950-61; PMID:11880364; http://dx.doi.org/10.1074/jbc.M108317200.
- Martin D, Galisteo R, Gutkind JS. CXCL8IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3Bcl10Malt1) complex. J Biol Chem 2009; 284:6038-42; PMID:19112107; http://dx.doi.org/10.1074/jbc.C800207200.
- Wang J, Guo Y, Zhang BC, Chen ZT, Gao JF. Induction of apoptosis and inhibition of cell migration and tube-like formation by dihydroartemisinin in murine lymphatic endothelial cells. Pharmacology 2007; 80:207-18; PMID:17622766; http://dx.doi.org/10.1159/000104418.
- Zhong YY, Zhang HF, Zhong JX, Bai L, Lu XH. Topical dihydroartemisinin inhibits suture-induced neovascularization in rat corneas through ERK12 and p38 pathways. Int J Ophthalmol 2011; 4:150-5; PMID:22553631.
- Gariano RF. Special features of human retinal angiogenesis. Eye (Lond) 2010; 24:401-7; PMID:20075971; http://dx.doi.org/10.1038/eye.2009.324.
- Grossniklaus HE, Kang SJ, Berglin L. Animal models of choroidal and retinal neovascularization. Prog Retin Eye Res 2010; 29:500-19; PMID:20488255; http://dx.doi.org/10.1016/j.preteyeres.2010.05.003.