
Abstract
Functional membrane androgen receptors (mAR) have previously been described in MCF-7 breast cancer cells. Their stimulation by specific testosterone albumin conjugates (TAC) activate rapidly non-genomic FAK/PI3K/Rac1/Cdc42 signaling, trigger actin reorganization and inhibit cell motility. PI3K stimulates serum and glucocorticoid inducible kinase SGK1, which in turn regulates the function of mAR. In the present study we addressed the role of SGK1 in mAR-induced apoptosis. TAC-stimulated mAR activation elicited apoptosis of MCF-7 cells, an effect significantly potentiated by concomitant incubation of the cells with TAC and the specific SGK1 inhibitors EMD638683 and GSK650394. In line with this, TAC and EMD638683 activated caspase-3. These effects were insensitive to the classical androgen receptor (iAR) antagonist flutamide, pointing to iAR-independent, mAR-induced responses. mAR activation and SGK1 inhibition further considerably augmented the radiation-induced apoptosis of MCF-7 cells. Moreover, TAC- and EMD638683 triggered early actin polymerization in MCF-7 cells. Blocking actin restructuring with cytochalasin B abrogated the TAC- and EMD638683-induced pro-apoptotic responses. Further analysis of the molecular signaling revealed late de-phosphorylation of FAK and Akt. Our results demonstrate that mAR activation triggers pro-apoptotic responses in breast tumor cells, an effect significantly enhanced by SGK1 inhibition, involving actin reorganization and paralleled by down-regulation of FAK/Akt signaling.
Abbreviations
| (mAR) | = | membrane androgen receptors |
| (TAC) | = | testosterone albumin conjugates |
| (SGK1) | = | serum and glucocorticoid inducible kinase |
| (iAR) | = | classical androgen receptor |
| (FAK) | = | Focal Adhesion Kinase |
Introduction
Expression of membrane androgen receptors (mARs) has been described in various tumor cells including prostate,Citation1-6 breast,Citation7,8 and colonCitation9,10 cancer as well as gliomas.Citation11 Their activation by specific membrane impermeable testosterone albumin conjugates (TAC) triggers various, classical androgen receptor (iAR)-independent non-genomic, signaling pathways that govern cellular outcomes including secretion, apoptosis and motility (for recent reviews seeCitation12,13). In particular, in MCF-7 breast cancer cells stimulation of functional mAR's was reported to activate rapidly the FAK/PI3K/Rac1/Cdc42 signaling that may trigger profound actin reorganization and significantly inhibit cell motility.Citation8 However, the initiation of mAR-induced pro-apoptotic responses in MCF-7 cells, as described in other tumor cell typesCitation7,9,14,15 has not been addressed.
Recently, we have reported that the serum and glucocorticoid inducible kinase SGK1 is implicated in the regulation of tumor growth in vivo, particularly in colonic tumors.Citation16,17 SGK1 facilitates cell survival mediated by the classical androgen receptor (iAR).Citation18,19 In addition, in a recent study we have shown that SGK1 may similarly regulate mAR functions.Citation20 Indeed, SGK1 was shown to enhance colon tumor cell migration via vinculin de-phosphorylation, a mechanism that was abrogated by mAR activation.Citation20 These findings imply a possible cross talk of SGK1 with membrane androgen receptors signaling and function. Since SGK1 is a downstream effector of PI3K implicated in pro-survival mechanismsCitation16 and PI3K is triggered by mAR stimulation in MCF-7 cellsCitation8 we addressed the possible role of SGK1 in mAR-initiated pro-apoptotic responses. In the present study we report for the first time a strong pro-apoptotic response to mAR stimulation that is significantly enhanced upon inhibition of SGK1 in mAR-stimulated MCF-7 cells. Moreover, combined SGK1 inhibition and mAR activation potentiates the radiation-induced apoptosis of MCF-7 cells. Our results indicate for the first time that mAR activation in combination with SGK1 inhibition and radiation may provide a powerful novel anti-tumorigenic strategy in the combat against breast cancer.
Results
TAC induces strong apoptotic response in MCF-7 human breast cancer cells that is enhanced by SGK1 inhibition
mAR expression has been previously reported in MCF-7 cells.Citation8 Here we first examined whether mAR activation induces apoptosis, as already described in prostate and colon tumor cells.Citation12 By using FACS analysis, we report strong pro-apoptotic response in MCF-7 cells treated for 24 h with 100 nM TAC (). In line with this finding caspase 3 was activated as early as 4 hours and up to 24 hours upon TAC stimulation of MCF-7 cells (). Since SGK1 is a pro-survival kinase, acting downstream of PI3K, we further examined whether the SGK1-specific inhibitors may influence the TAC-induced breast tumor growth control. For this we used EMD638683 (50 μM) and GSK650394 (1μM) that are selective SGK inhibitors and EMD638683 is highly effective in vivo.Citation21 FACS analysis revealed that combined treatment of MCF-7 cells with both the SGK1 inhibitors and TAC significantly enhanced the TAC-induced apoptotic response (), reaching a maximum rate of 50% after combined treatment. Moreover, results shown in indicate that early apoptosis was significantly higher than late apoptosis. Whether this finding may reflect a partial post-treatment recovery of the cells or experimental limitations in the treatment efficacy remain to be elucidated. SGK1 inhibitors in combination with TAC significantly up-regulated caspase 3 activity further (). These findings indicate that SGK1 inhibition interferes with mAR-induced signaling and function and potentiates the apoptotic response in MCF-7 breast cancer cells. We have further addressed the possible involvement of the classical intracellular androgen receptor (iAR) in the observed TAC effects on MCF-7 cells. For this, both, apoptosis and caspase 3 activity were measured in the presence of the androgen receptor antagonist flutamide. As shown in the apoptotic responses and the activation of caspase 3 remained unchanged under all experimental conditions in the presence of 1 μM flutamide, implying an iAR-independent, mAR-induced effect.
Figure 1. Early and late apoptosis of MCF-7 breast cancer cells treated by TAC and SGK1 inhibitors EMD638683 and GSK650394. (A) Original dot-plots (PI/Annexin V) of a representative expriment demonstrating an increase of cell events in the lower right quadrant (early apoptosis) and upper right quadrant (late apoptosis) in TAC- and TAC+EMD638683- or TAC+GSK650394- treated group in the presence and absence of 1 μM flutamide. (B,C) Flow cytometry results after a 24h treatment in the absence (control) or presence of 100 nM TAC, 50 μM EMD638683, combination of TAC+EMD638683, in the presence or absence of 1 μM flutamide, or 1 μl solvent (DMSO) following staining with FITC conjugated Annexin V and propidium iodide (PtdIns). Presented results are arithmetic means ± SEM (n = 6) of the percentage gated MCF-7 cells binding Annexin V but not propidium iodide (early apoptosis, B), or both Annexin V and propidium iodide (late apoptosis, C). * (P < 0 .05), ** (P < 0 .01) and *** (P < 0.001) indicate significant differences to respective value of untreated control. &&& (P < 0 .01) indicates significant differences of TAC+EMD638683-treated cells to respective values of TAC-treated cells (unpaired t-test). Note that no differences are observed between presence or absence of 1 μM flutamide.
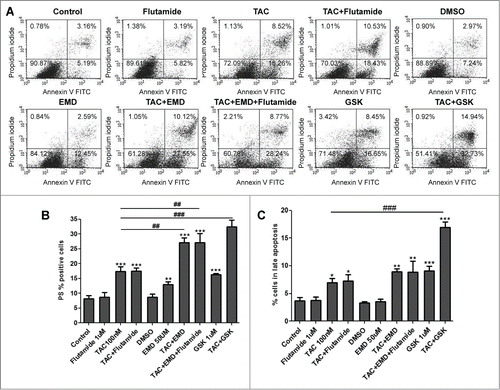
Figure 2. Caspase 3 activation of MCF-7 breast cancer cells treated by TAC and SGK1 inhibitor EMD638683. After 2, 4, 6, and 24h exposure in the absence (control) or presence of 100 nM TAC, 50 μM EMD638683 or TAC+EMD638683 in the presence or absence of 1 μM flutamide, or 1 μl solvent (DMSO), cells were stained with conjugated inhibitor of active Caspase-3 (FITC-DEVD-FMK) and measured by FACS. Graph is showing arithmetic means ± SEM (n = 6) of the percentage gated MCF-7 cells. ** (P < 0 .01) and *** (P < 0.001) indicate significant differences to respective value of untreated control (unpaired t-test). ## (P < 0 .01) indicates significant differences of TAC+EMD638683 treated cells to respective values of TAC-treated cells. Note that no differences are observed between presence or absence of 1 μM flutamide.
mAR activation and SGK1 inhibition enhance radiation-induced cell growth control
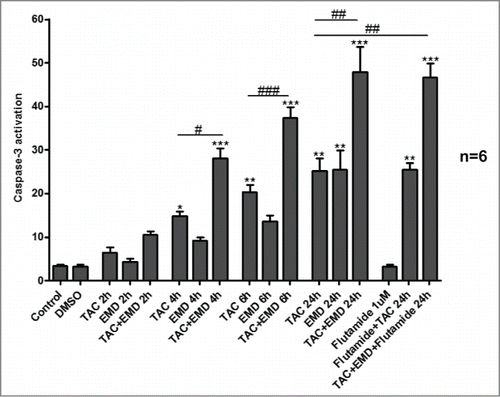
Since radiation is widely used in human tumor treatment, we further analyzed whether mAR activation and SGK1 inhibition may interfere with radiation-induced cell death. For this, MCF-7 cells were treated for 24 h with TAC (100 nM), EMD638683 (50 μM) or the combination of both and then irradiated or not (as described in materials and methods). Apoptosis was assessed following incubation for additional 72 h. As shown in the radiation-induced early and late apoptosis was significantly enhanced in cells pretreated with TAC or EMD638683, while combined treatment of the irradiated cells with both agents further potentiated the pro-apoptotic responses.
Figure 3. SGK1 inhibitor EMD638683 enhances radiation-induced apoptosis in TAC-treated MCF-7 cells. Original dot-plots (PI/Annexin V) of a representative expriment demonstrating an increase of cell events in the lower right quadrant (early apoptosis) and upper right quadrant (late apoptosis) in (A) non-irradiated TAC- and TAC+EMD638683-treated cells and (B) 4 Gy irradiated TAC- and TAC+EMD638683-treated MCF-7 cells. (C,D) Flow cytometry results after a 24h treatment in the absence (control) or presence of 100 nM TAC, 50 μM EMD638683, combination of TAC+EMD638683, or 1 μl solvent (DMSO) in non-irradiated- (white bars) or 4 Gy irradiated- (black bars) MCF-7 cells following staining with FITC conjugated Annexin V and propidium iodide (PtdIns). Results represent arithmetic means ± SEM (n = 6) of the percentage gated MCF7 cells binding to Annexin V but not propidium iodide (early apoptosis, C), and to both Annexin V and propidium iodide (late apoptosis, D). ** (P < 0 .01) and *** (P < 0,001) indicate significant differences to respective values of untreated control. # (P < 0 .05), ## (P < 0 .01) and ### (P < 0 .001) indicate significant differences of irradiated cells to respective values of non-irradiated cells. $ (P < 0 .05), $$ (P < 0 .01) and $$$ (P < 0 .001) indicate significant differences to respective values of irradiated control. &&& (P < 0 .001) indicates significant difference of irradiated TAC+EMD638683-treated cells to respective values of irradiated TAC-treated cells (unpaired t-test). § (P < 0 .05), indicates significance between TAC-treated and TAC+END-treated non-irradiated cells.
Actin redistribution controls the mAR and SGK1-governed pro-apoptotic responses
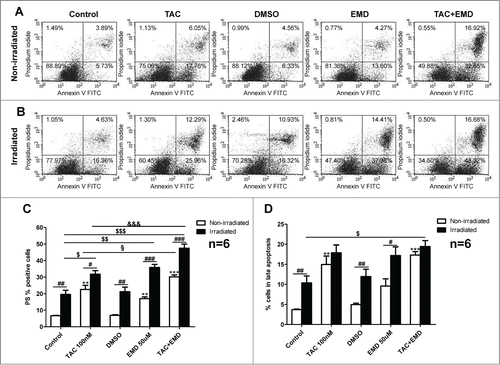
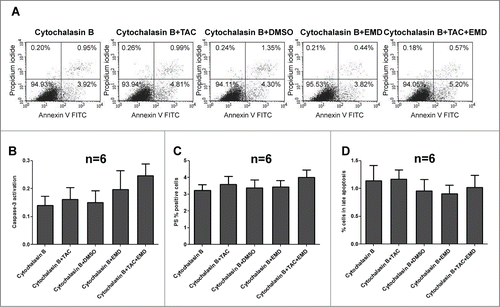
Previous studies revealed that early actin reorganization regulated by mAR activation govern the TAC-induced pro-apoptotic responses in prostateCitation2,14 and colonCitation9 cancer cells. Moreover, actin redistribution has been reported to be a prominent mAR response in MCF-7 cells.Citation8 In line with these reports, strong and transient actin polymerization was evident in MCF-7 cells upon mAR activation as documented by the significant decrease of the G/F-actin ratio (). This effect was manifested as early as 15 minutes and persisted for 60 minutes upon TAC treatment. Interestingly, inhibition of SGK1 by EMD638683 induced in addition rapid and transient actin polymerization (), implying that SGK1 signaling is involved in the reorganization of actin architecture in breast cancer cells. We further examined whether actin remodeling is involved in the mAR- and SGK1-regulated apoptosis of MCF-7 cells. For this, cells were pretreated with the actin cytoskeleton inhibitor cytochalasin B (1 μM) that blocks actin polymerization prior to the stimulation of mAR by TAC and inhibition of SGK1 by EMD638683. The estimation of early and late apoptosis was followed by FACS analysis. As shown in the pro-apoptotic effects were clearly inhibited, while caspase 3 activation () was abolished, indicating that actin restructuring is a crucial step involved in the pro-apoptotic signaling mechanism of MCF-7 breast cancer cells.
Figure 4. Effects of TAC and TAC+SGK1 inhibitor EMD638683 in actin polymerization dynamics in MCF-7 cells. Arithmetic means ± SEM (n = 4 independent experiments) of G/F actin ratio (normalized) in MCF-7 breast cancer cells without or with prior treatment with 100 nM TAC, 50 μM EMD638683, or combined TAC+EMD638683 for 15, 30 and 60 minutes. * (P < 0 .05), ** (P < 0 .01) and *** (P < 0 .001) indicates statistically significant differences to respective value of untreated control. # (P < 0 .05) indicates significant differences of TAC+EMD638683 treated cells to respective values of TAC-treated cells.
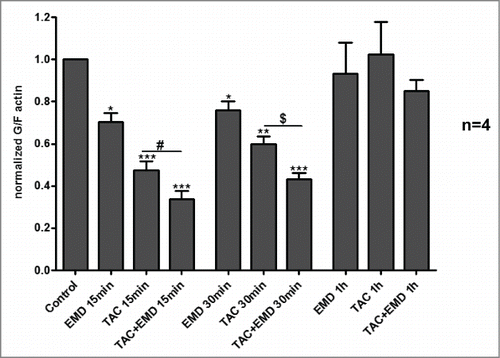
Figure 5. Cytochalasin (B)inhibits the pro-apoptotic effects in TAC-, EMD638683- and TAC+EMD638683-treated MCF-7 cells. Original dot-plots (PI/Annexin V) (A) of a representative expriment demonstrating TAC-, EMD638683 and TAC+EMD638683-treated MCF7 cells in the presence of 1 μM cytochalasin B. Flow cytometry results after a 24h treatment in the presence of 1 μM cytochalasin B of MCF-7 cells treated with 100 nM TAC, 50 μM EMD638683 or TAC+EMD638683 following staining with conjugated inhibitor of active Caspase-3 (FITC-DEVD-FMK) (B), with FITC conjugated Annexin V (C) and propidium iodide (PtdIns) (D). Presented results are arithmetic means ± SEM (n = 6). Note that no significant effects of TAC and EMD638683-treatment were observed in the presence of 1 μM cytochalasin B.
mAR activation and SGK1 inhibition induce late FAK and Akt dephosphorylation in MCF-7 cells
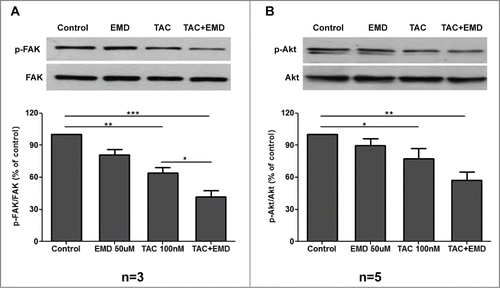
We have further addressed the identification of possible signaling molecules that may be implicated in mAR-induced pro-apoptotic responses regulated by SGK1 in MCF-7 cells. For this we assessed the phosphorylation state of the Focal Adhesion Kinase FAK. FAK is important for normal cell survival maintenance, while disruption of FAK signaling initiates loss of substrate adhesion and apoptotic responses.Citation22 As shown in , the ratio of phosphorylated FAK to the total FAK protein decreased significantly after long term (24 h) incubation of the cells with TAC, or TAC plus EMD638683 respectively. FAK signaling may activate downstream molecules such as PI3 K/Akt.Citation22,23 For this we further analyzed the phosphorylation state of Akt. In line with the observed FAK dephosphorylation a significant decrease of the ratio of phosphorylated Akt to total protein content became evident in MCF-7 cells, incubated for 24 hours with TAC or TAC plus EMD638683 (). Taken together these results imply the long-term downregulation of a central pro-survival signaling pathway in mAR and SGK1-governed pro-apoptotic responses in MCF-7 cells.
Figure 6. Effects of TAC and TAC+SGK1 inhibitor EMD638683 on FAK and Akt phosphorylation in MCF-7 cells. (A) MCF-7 cells were stimulated with 100 nM TAC, 50 μM EMD638683 or combined TAC+EMD638683 for 24 h. Following cell lysis equal amounts of total lysates were immunoblotted (IB) with a specific antibody against phospho-FAK and total FAK. Immunoblots were analyzed by densitometry. The intensity of phospho-FAK was normalized to the intensity of the corresponding total FAK band. Blots are from a representative experiment, whereas the relative fold decrease are indicated as mean values ± SEM from n = 3 independent experiments with that of untreated cells taken as 1 (*P < 0.05; **P < 0.01 ***P < 0.001). (B) MCF-7 cells were stimulated with 100 nM TAC, 50 μM EMD638683 or combined TAC+EMD638683 for 24h. Following cell lysis equal amounts of total lysates were immunoblotted (IB) with a specific antibody against phospho-Akt and total Akt. Immunoblots were analyzed by densitometry. The intensity of phospho-Akt was normalized to the intensity of the corresponding total Akt band. Blots are from a representative experiment, whereas the relative fold decrease are indicated as mean values ± SEM from n = 5 independent experiments with that of untreated cells taken as 1 (*P < 0.05; **P < 0.01).
Discussion
The present study discloses the novel finding that mAR activation by testosterone albumin conjugates along with SGK1 inhibition triggers strong pro-apoptotic responses in MCF-7 human breast cancer cells. The significant role of SGK1 in the mAR-induced apoptotic response was established for the first time, and shown to be enhanced substantially by the specific SGK1 inhibitors EMD638683 and GSK650394. Interestingly, using the anti-androgen flutamide alone or in combination with TAC or EMD did not impact on cell fate, indicating that the classical intracellular AR may not be involved in the membrane androgen receptor actions. Moreover, we report that mAR activation and SGK1 inhibition in combination to radiotherapy further potentiated the radiation-induced apoptosis of MCF-7 cells. These findings prove a functional cross-talk of mAR activation with SGK1 signaling that may govern pro-apoptotic responses in breast tumor cells.
SGK1 is a family member of the SGK pro-survival kinases acting downstream of PI3 K, implicated in tumor growth.Citation16,17,24,25 SGK1 is also involved in the signaling of migration.Citation26 In addition, SGK1-dependent Ca2+ signaling was recently proposed as a novel mechanism regulating the actin cytoskeleton in mast cells.Citation27 Our findings showing reorganization of actin architecture in breast cancer cells upon SGK1 inhibition fully supports this hypothesis. The possible crosstalk of SGK1 with membrane testosterone signaling and actin reorganization has been recently addressed.Citation20 It was shown that the enhanced migratory and invasive potential of colon tumor cells expressing the active mutant of SGK1 was strongly inhibited upon stimulation of membrane androgen receptors by testosterone and testosterone conjugates.Citation20 These results imply a significant role of this pro-survival kinase acting downstream of PI3K in membrane-initiated testosterone actions. Although a direct short term phosphorylation of endogenous SGK1 upon mAR stimulation was not observed in MCF7 cells (data not shown), it is reasonable to assume that the inhibition of SGK1 potentiates the mAR-dependent pro-apoptotic effect as a result of the inhibition of this important pro-survival factor.
An additional novel finding of the present work is the observed strong enhancement of the pro-apoptotic responses after combined treatment of irradiated MCF-7 cells with both agents, reaching a maximal 65% apoptotic rate that may reflect the margins in the treatment efficacy of this cell line. These findings suggest that mAR activation by TAC and SGK1 inhibition in combination to radiation may substantially improve tumor regression in human breast cancer cells and further support recent findings on EMD638683 action in colon tumor cells.Citation28 However, since existing reports so far do not support the role of apoptosis in radiation-induced clonogenic inactivity of solid tumors,Citation29-31 further studies are now needed to analyze in detail the clinical significance of these interactions.
Recent observations have shown that actin reorganization initiated by androgens is controlling various cellular functions including apoptosis in mAR-stimulated human prostate and colon cancer cells.Citation9,10,14 The role of actin reorganization is underscored by our present observations, showing clear inhibition of the mAR and SGK1-governed apoptotic response in breast cancer cells pre-treated with cytochalasin B that blocks the observed actin reorganization. Taken together these findings suggest a pivotal role of actin reorganization that may link membrane-initiated steroid hormone actions to the regulation of cellular responses in tumor cells and further support the recently postulated key role of actin remodeling in apoptosis, cell death and aging.Citation32-35 However, despite various molecular mechanisms that have been proposed to establish a link between early actin redistribution and late cellular outcomes,Citation14,34-37 the molecular basis of these interactions is still not well understood (for a recent review see Citation38).
Our present findings further revealed late down-regulation of FAK and Akt signaling upon mAR stimulation and SGK1 inhibition in breast cancer cells. Although late FAK and Akt activation status may not provide direct mechanistic insights, these results provide additional molecular indications supporting the pro-apoptotic response of the cells that could be partially correlated with the molecular signaling governing cell survival and motility responses, as this has been previously reported in various tumor cells.Citation23 Further work is now needed to address this important issue.
Conclusions
In the present study we report for the first time that SGK1 is involved in membrane-initiated testosterone pro-apoptotic actions in tumor cells. Inhibition of SGK1 by specific inhibitors enhances mAR-induced apoptosis in MCF-7 breast cancer cells. This interaction involves early actin reorganization and late FAK and Akt inactivation. In addition, mAR activation and concomitant SGK1 inhibition potentiates considerably the radiation-induced cell death of breast tumor cells. These findings imply that mAR activation in combination with SGK1 inhibition and radiation may offer a novel anti-tumorigenic option in the treatment of breast cancer.
Materials and Methods
Cell cultures
MCF-7 mammary adenocarcinoma cells, provided from ATCC were cultured in 1:1 Dulbecco's Modified Eagle Medium (DMEM)/Ham's F12 medium supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 30 mM NaHCO3, 16 ng/ml insulin and 50 mg/ml penicillin/streptomycin. Cells were maintained in a humidified atmosphere of 5%, CO2 in 25 cm2 or 75 cm2 flasks. Subcultivation was performed with Ca2+ and Mg2+ free phosphate buffer saline (PBS) containing 0.25% trypsin and 5 mM EDTA. Based on previous titration experiments,Citation3,8,9 for mAR stimulation we have used throughout this study the non-permeable androgen derivative testosterone-BSA (TAC, Sigma-Aldrich) in a concentration of 100 nM. In distinct experiments, the anti-androgen drug flutamide (1 μM, Sigma), the SGK1-specific inhibitors EMD638683 (50 μM) and GSK650394 (1 μM), as well as the actin cytoskeleton-disrupting agent cytochalasin B (1 μM, Sigma-Aldrich) were used as indicated.
Irradiation of MCF-7 cells
Irradiation was performed at 37°C using a Gulmay RS225 X-ray machine (Gulmay limited, Chertsey, UK) with a dose rate of 1.0 Gy/minute and the exposure factors of 200 kVp, 15 mA and 0.5-mm Copper additional filtering. 2.0 × 105 cells were seeded in 6-well plates and cultured with fresh culture medium for 24 h, after which 100 nM testosterone-BSA was applied in the presence or absence of EMD638683 (50 μM) for 24h. For comparison, the cells were treated with the solvent DMSO and one solvent control was analyzed with each set of experiments. The cells were subsequently exposed to 4 Gy. After further incubation for 72h the apoptosis of cells was analyzed utilizing flow cytometry (BD FACS Calibur, BD Biosciences).
FACS analysis
Apoptosis assay
The quantification of apoptotic cells was estimated by flow cytometry using the Annexin-V-FITC/Propidium iodide (PI) apoptosis detection kit in accordance with the manufacturer's instructions (Mabtag). Briefly, 2.0 × 105 cells were seeded into 6-well plates and after indicated treatment, the cells were harvested, washed with cold PBS and then suspended in annexin V binding buffer containing annexin V-FITC solution and propidium iodide (PtdIns). The cells were incubated in the dark for 20 minutes at room temperature and then measured immediately yielding a FL-1 vs FL-2 dot plot in FACS (BD FACS Calibur, BD Biosciences).
Caspase-3 activation
Active caspase-3 was measured by CaspGlow Fluorescein Active Caspase-3 Staining kit from BioVision adhering to the manufacturer's instruction. Briefly, 1.0 × 105 cells were suspended in 100 μl complete DMEM, stained for 1 hour with 0.2 μl active caspase-3 FITC antibody under cell culture conditions. The cells were then washed once in 200 μl wash buffer supplied in the kit and resuspended in 200 μl wash buffer for immediate FACS analysis.
Estimate of actin cytoskeleton dynamics
MCF-7 cells were incubated with 100 nM testosterone-BSA in the presence or absence of 50 μM EMD638683 for the indicated time periods. The actin cytoskeleton dynamics were estimated by FACS analysis as described previously.Citation39 In brief, cells were fixed with paraformaldehyde (4%, 15 min, 20°C), washed 3 times with PBS, permeabilized with 0.1% Triton X-100 (5 min, 4°C), washed 3 times with PBS plus 0.1% FCS, and then stained with 5 μg/ml DNaseI Alexa-488 and phalloidin Alexa-647 (1:100) (Molecular Probes). The G-actin/F-actin ratio was calculated using the mean fluorescent intensity measured by FACS. Decrease of this ratio indicates actin polymerization.Citation40,41
Western blotting
Cells were incubated with 100 nM testosterone-BSA in the presence or absence of 50 μM EMD638683 for 24 h, washed twice with ice-cold PBS and suspended in ice-cold lysis buffer (50 mM Tris/HCl, 1% TritonX-100 pH 7.4, 1% sodium deoxycholate, 0.1% SDS, 0.15% NaCl, 1 mM EDTA, 1 mM sodium orthovanadate) containing a protease inhibitor cocktail (Sigma). The protein concentration was determined using the Bradford assay (BioRad). Sixty μg of protein were solubilized in sample buffer at 95°C for 5 min and resolved by 10% SDS-PAGE. For immunoblotting proteins were electro-transferred onto a PVDF membrane and blocked with 5% nonfat milk in TBS-0.10% Tween 20 at room temperature for 1 h. Then, the membrane was incubated with anti-phospho-FAK (Tyr397; 1:1000, Cell Signaling), anti-FAK (1:1000, Cell Signaling), anti-phospho-Akt (Thr308; 1:1000, Cell Signaling), anti-Akt (1:1000, Cell Signaling) at 4°C overnight. After washing (TBST) and subsequent blocking the blot was incubated with secondary anti rabbit antibody (1:2000, Cell Signaling) for 1 h at room temperature. After washing, antibody binding was detected with the ECL detection reagent (Amersham).
Statistical analysis
All results are presented as means ± SEM, while n represents the number of independent experiments. Significance was assessed using unpaired student's t-test or ANOVA as appropriate. p-values < 0.05 were considered statistically significant. Statistical analysis was performed with GraphPad InStat version 3.00 for Windows 95, GraphPad Software, www.graphpad.com.
Authors’ Contributions
GL and GL carried out the molecular studies and FACS analysis. SH and SS performed the cellular studies and Western blotting. SP and SA carried out the actin cytoskeleton determinations. MT participated in the radiation experiments and interpretation of the data. FL and CS drafted the manuscript. CS conceived of the study, and participated with FL in its design and coordination. All authors read and approved the final manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors gratefully acknowledge the meticulous preparation of the manuscript by Tanja Loch.
Funding
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB773), the University of Crete Research Committee (KA 3452), and the Deanship of Scientific Research at King Saud University (KSU-RGP-018 and Programs).
References
- Kampa M, Papakonstanti EA, Hatzoglou A, Stathopoulos EN, Stournaras C, Castanas E. The human prostate cancer cell line LNCaP bears functional membrane testosterone receptors that increase PSA secretion and modify actin cytoskeleton. FASEB J 2002; 16:1429-31; PMID:12205037
- Papadopoulou N, Charalampopoulos I, Alevizopoulos K, Gravanis A, Stournaras C. Rho/ROCK/actin signaling regulates membrane androgen receptor induced apoptosis in prostate cancer cells. Exp Cell Res 2008; 314:3162-74; PMID:18694745; http://dx.doi.org/10.1016/j.yexcr.2008.07.012
- Papakonstanti EA, Kampa M, Castanas E, Stournaras C. A rapid, nongenomic, signaling pathway regulates the actin reorganization induced by activation of membrane testosterone receptors. Mol Endocrinol 2003; 17:870-81; PMID:12554777; http://dx.doi.org/10.1210/me.2002-0253
- Sen A, O’Malley K, Wang Z, Raj GV, Defranco DB, Hammes SR. Paxillin regulates androgen- and epidermal growth factor-induced MAPK signaling and cell proliferation in prostate cancer cells. J Biol Chem 2010; 285:28787-95; PMID:20628053; http://dx.doi.org/10.1074/jbc.M110.134064
- Sun YH, Gao X, Tang YJ, Xu CL, Wang LH. Androgens induce increases in intracellular calcium via a G protein-coupled receptor in LNCaP prostate cancer cells. J Androl 2006; 27:671-8; PMID:16728719; http://dx.doi.org/10.2164/jandrol.106.000554
- Kampa M, Kogia C, Theodoropoulos PA, Anezinis P, Charalampopoulos I, Papakonstanti EA, Stathopoulos EN, Hatzoglou A, Stournaras C, Gravanis A, et al. Activation of membrane androgen receptors potentiates the antiproliferative effects of paclitaxel on human prostate cancer cells. Mol Cancer Ther 2006; 5:1342-51; PMID:16731768; http://dx.doi.org/10.1158/1535-7163.MCT-05-0527
- Kampa M, Nifli AP, Charalampopoulos I, Alexaki VI, Theodoropoulos PA, Stathopoulos EN, Gravanis A, Castanas E. Opposing effects of estradiol- and testosterone-membrane binding sites on T47D breast cancer cell apoptosis. Exp Cell Res 2005; 307:41-51; PMID:15922725; http://dx.doi.org/10.1016/j.yexcr.2005.02.027
- Kallergi G, Agelaki S, Markomanolaki H, Georgoulias V, Stournaras C. Activation of FAK/PI3K/Rac1 signaling controls actin reorganization and inhibits cell motility in human cancer cells. Cell Physiol Biochem 2007; 20:977-86; PMID:17982280; http://dx.doi.org/10.1159/000110458
- Gu S, Papadopoulou N, Gehring EM, Nasir O, Dimas K, Bhavsar SK, Föller M, Alevizopoulos K, Lang F, Stournaras C. Functional membrane androgen receptors in colon tumors trigger pro-apoptotic responses in vitro and reduce drastically tumor incidence in vivo. Mol Cancer 2009; 8:114; PMID:19948074; http://dx.doi.org/10.1186/1476-4598-8-114
- Gu S, Papadopoulou N, Nasir O, Foller M, Alevizopoulos K, Lang F, Stournaras C. Activation of membrane androgen receptors in colon cancer inhibits the prosurvival signals Akt/bad in vitro and in vivo and blocks migration via vinculin/actin signaling. Mol Med 2011; 17:48-58; PMID:20957335; http://dx.doi.org/10.2119/molmed.2010.00120
- Gatson JW, Kaur P, Singh M. Dihydrotestosterone differentially modulates the mitogen-activated protein kinase and the phosphoinositide 3-kinase/Akt pathways through the nuclear and novel membrane androgen receptor in C6 cells. Endocrinology 2006; 147:2028-34; PMID:16410299; http://dx.doi.org/10.1210/en.2005-1395
- Lang F, Alevizopoulos K, Stournaras C. Targeting membrane androgen receptors in tumors. Expert Opin Ther Targets 2013; 17:951-63; PMID:23746222; http://dx.doi.org/10.1517/14728222.2013.806491
- Papadopoulou N, Papakonstanti EA, Kallergi G, Alevizopoulos K, Stournaras C. Membrane androgen receptor activation in prostate and breast tumor cells: molecular signaling and clinical impact. IUBMB Life 2009; 61:56-61; PMID:19109827; http://dx.doi.org/10.1002/iub.150
- Papadopoulou N, Charalampopoulos I, Anagnostopoulou V, Konstantinidis G, Foller M, Gravanis A, Alevizopoulos K, Lang F, Stournaras C. Membrane androgen receptor activation triggers down-regulation of PI-3K/Akt/NF-kappaB activity and induces apoptotic responses via Bad, FasL and caspase-3 in DU145 prostate cancer cells. Molecular cancer 2008; 7:88; PMID:19055752; http://dx.doi.org/10.1186/1476-4598-7-88
- Hatzoglou A, Kampa M, Kogia C, Charalampopoulos I, Theodoropoulos PA, Anezinis P, Dambaki C, Papakonstanti EA, Stathopoulos EN, Stournaras C, et al. Membrane androgen receptor activation induces apoptotic regression of human prostate cancer cells in vitro and in vivo. J Clin Endocrinol Metab 2005; 90:893-903; PMID:15585562; http://dx.doi.org/10.1210/jc.2004-0801
- Lang F, Perrotti N, Stournaras C. Colorectal carcinoma cells-regulation of survival and growth by SGK1. Int J Biochem Cell Biol 2010; 42:1571-5; PMID:20541034; http://dx.doi.org/10.1016/j.biocel.2010.05.016
- Lang F, Stournaras C. Serum and glucocorticoid inducible kinase, metabolic syndrome, inflammation, and tumor growth. Hormones 2013; 12:160-71; PMID:23933686; http://dx.doi.org/10.14310/horm.2002.1401
- Shanmugam I, Cheng G, Terranova PF, Thrasher JB, Thomas CP, Li B. Serum/glucocorticoid-induced protein kinase-1 facilitates androgen receptor-dependent cell survival. Cell Death Differ 2007; 14:2085-94; PMID:17932503; http://dx.doi.org/10.1038/sj.cdd.4402227
- Zou JX, Guo L, Revenko AS, Tepper CG, Gemo AT, Kung HJ, Chen HW. Androgen-induced coactivator ANCCA mediates specific androgen receptor signaling in prostate cancer. Cancer Res 2009; 69:3339-46; PMID:19318566; http://dx.doi.org/10.1158/0008-5472.CAN-08-3440
- Schmidt EM, Gu S, Anagnostopoulou V, Alevizopoulos K, Foller M, Lang F, Stournaras C. Serum- and glucocorticoid-dependent kinase-1-induced cell migration is dependent on vinculin and regulated by the membrane androgen receptor. FEBS J 2012; 279:1231-42; PMID:22309306; http://dx.doi.org/10.1111/j.1742-4658.2012.08515.x
- Ackermann TF, Boini KM, Beier N, Scholz W, Fuchss T, Lang F. EMD638683, a novel SGK inhibitor with antihypertensive potency. Cell Physiol Biochem 2011; 28:137-46; PMID:21865856; http://dx.doi.org/10.1159/000331722
- Lu Q, Rounds S. Focal adhesion kinase and endothelial cell apoptosis. Microvasc Res 2012; 83:56-63; PMID:21624380; http://dx.doi.org/10.1016/j.mvr.2011.05.003
- Hanks SK, Ryzhova L, Shin NY, Brabek J. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front Biosci 2003; 8:d982-96; PMID:12700132; http://dx.doi.org/10.2741/1114
- Lang F, Voelkl J. Therapeutic potential of serum and glucocorticoid inducible kinase inhibition. Expert Opin Investig Drugs 2013; 22:701-14; PMID:23506284; http://dx.doi.org/10.1517/13543784.2013.778971
- Nasir O, Wang K, Foller M, Gu S, Bhandaru M, Ackermann TF, Boini KM, Mack A, Klingel K, Amato R, et al. Relative resistance of SGK1 knockout mice against chemical carcinogenesis. IUBMB Life 2009; 61:768-76; PMID:19548318; http://dx.doi.org/10.1002/iub.209
- Schmidt EM, Kraemer BF, Borst O, Munzer P, Schonberger T, Schmidt C, Leibrock C, Towhid ST, Seizer P, Kuhl D, et al. SGK1 sensitivity of platelet migration. Cell Physiol Biochem 2012; 30:259-68; PMID:22759972; http://dx.doi.org/10.1159/000339062
- Schmid E, Gu S, Yang W, Munzer P, Schaller M, Lang F, Stournaras C, Shumilina E. Serum- and glucocorticoid-inducible kinase SGK1 regulates reorganization of actin cytoskeleton in mast cells upon degranulation. Am J Physiol Cell Physiol 2013; 304:C49-55; PMID:23015548; http://dx.doi.org/10.1152/ajpcell.00179.2012
- Towhid ST, Liu GL, Ackermann TF, Beier N, Scholz W, Fuchss T, Toulany M, Rodemann HP, Lang F. Inhibition of colonic tumor growth by the selective SGK inhibitor EMD638683. Cell Physiol Biochem 2013; 32:838-48; PMID:24081014; http://dx.doi.org/10.1159/000354486
- Toulany M, Kehlbach R, Florczak U, Sak A, Wang S, Chen J, Lobrich M, Rodemann HP. Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Mol Cancer Ther 2008; 7:1772-81; PMID:18644989; http://dx.doi.org/10.1158/1535-7163.MCT-07-2200
- Brown JM, Wouters BG. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res 1999; 59:1391-9; PMID:10197600
- Kasten-Pisula U, Windhorst S, Dahm-Daphi J, Mayr G, Dikomey E. Radiosensitization of tumour cell lines by the polyphenol Gossypol results from depressed double-strand break repair and not from enhanced apoptosis. Radiother Oncol 2007; 83:296-303; PMID:17521756; http://dx.doi.org/10.1016/j.radonc.2007.04.024
- Franklin-Tong VE, Gourlay CW. A role for actin in regulating apoptosis/programmed cell death: evidence spanning yeast, plants and animals. Biochem J 2008; 413:389-404; PMID:18613816; http://dx.doi.org/10.1042/BJ20080320
- Gourlay CW, Ayscough KR. The actin cytoskeleton: a key regulator of apoptosis and ageing? Nat Rev Mol Cell Biol 2005; 6:583-9; PMID:16072039; http://dx.doi.org/10.1038/nrm1682
- Papakonstanti EA, Stournaras C. Tumor necrosis factor-alpha promotes survival of opossum kidney cells via Cdc42-induced phospholipase C-gamma1 activation and actin filament redistribution. Mol Biol Cell 2004; 15:1273-86; PMID:14699068; http://dx.doi.org/10.1091/mbc.E03-07-0491
- Wang Y, George SP, Srinivasan K, Patnaik S, Khurana S. Actin reorganization as the molecular basis for the regulation of apoptosis in gastrointestinal epithelial cells. Cell Death Differ 2012; 19:1514-24; PMID:22421965; http://dx.doi.org/10.1038/cdd.2012.28
- Kramar EA, Chen LY, Brandon NJ, Rex CS, Liu F, Gall CM, Lynch G. Cytoskeletal changes underlie estrogen's acute effects on synaptic transmission and plasticity. J Neurosci 2009; 29:12982-93; PMID:19828812; http://dx.doi.org/10.1523/JNEUROSCI.3059-09.2009
- Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 2010; 11:353-65; PMID:20414257; http://dx.doi.org/10.1038/nrm2890
- Stournaras C, Gravanis A, Margioris AN, Lang F. The actin cytoskeleton in rapid steroid hormone actions. Cytoskeleton 2014; 71:285-93; PMID:24616288; http://dx.doi.org/10.1002/cm.21172
- Zaru R, Mollahan P, Watts C. 3-phosphoinositide-dependent kinase 1 deficiency perturbs Toll-like receptor signaling events and actin cytoskeleton dynamics in dendritic cells. J Biol Chem 2008; 283:929-39; PMID:17991746; http://dx.doi.org/10.1074/jbc.M708069200
- Gu S, Kounenidakis M, Schmidt EM, Deshpande D, Alkahtani S, Alarifi S, Föller M, Alevizopoulos K, Lang F, Stournaras C. Rapid activation of FAK/mTOR/p70S6K/PAK1-signaling controls the early testosterone-induced actin reorganization in colon cancer cells. Cell Signal 2013; 25:66-73; PMID:23316499; http://dx.doi.org/10.1016/j.cellsig.2012.08.005
- Papakonstanti EA, Stournaras C. Actin cytoskeleton architecture and signaling in osmosensing. Methods Enzymol 2007; 428:227-40; PMID:17875420; http://dx.doi.org/10.1016/S0076-6879(07)28012-7