
Abstract
Multidrug resistance (MDR) is one of the major obstacles to the efficiency of cancer chemotherapy, which often results from the overexpression of drug efflux transporters such as P-glycoprotein (P-gp). In the present study, we determined the effect of dasatinib which was approved for imatinib resistant chronic myelogenous leukemia (CML) and (Ph+) acute lymphoblastic leukemia (ALL) treatment on P-gp-mediated MDR. Our results showed that dasatinib significantly increased the sensitivity of P-gp-overexpressing MCF-7/Adr cells to doxorubicin in MTT assays; thus lead to an enhanced cytotoxicity of doxorubicin in MCF-7/Adr cells. Additionally, dasatinib increased the intracellular accumulation, inhibited the efflux of doxorubicin in MCF-7/Adr cells, and significantly enhanced doxorubicin-induced apoptosis in MCF-7/Adr cells. Further studies showed that dasatinib altered the expression levels of mRNA, protein levels of P-gp, and the phosphorylation of signal–regulated kinase (ERK) both in time-dependent (before 24 h) and dose-dependent manners at concentrations that produced MDR reversals. In conclusion, dasatinib reverses P-gp-mediated MDR by downregulating P-gp expression, which may be partly attributed to the inhibition of ERK pathway. Dasatinib may play an important role in circumventing MDR when combined with other conventional antineoplastic drugs.
Abbreviations
| MDR | = | multidrug resistance |
| P-gp | = | P-glycoprotein |
| DOX | = | doxorubicin |
| = | ERKextracellular signal-regulated kinase | |
| P-ERK | = | phosphorylated extracellular signal–regulated kinase |
Introduction
Multidrug resistance (MDR) causes resistance of the cytotoxic effects of numerous antitumor drugs that are structurally and mechanistically unrelated, thus significantly decreases the efficacy of cancer chemotherapy. The mechanisms of MDR vary but the most common cause is the overexpression of cell membrane-bound ATP-binding cassette (ABC) transporters. These transporters coupled with energy consumption pump out exogenous toxic substances or metabolite from the inside of cells. Therefore, the effluxes of various chemotherapeutic drugs lead to attenuation of their cytotoxic effects on tumor cells. Among the well-known 48 ABC proteins, P-glycoprotein (P-gp) or ABC subfamily B member 1 (ABCB1), multidrug resistance-associated protein 1 (MRP1) or ABC subfamily C member 1 (ABCC1), and breast cancer resistance protein (BCRP) or ABC subfamily G member 2 (ABCG2) all play significant roles in causing MDR in cancer cells.Citation1
P-gp, a 170–180 kDa membrane glycoprotein, was first discovered in drug-resistant Chinese hamster ovarian cells.Citation2 As an ATP dependent drug efflux pump, it hydrolyses ATP to efflux a broad spectrum of hydrophobic agents that includes lipids, bile acids, xenobiotics and peptides. It also preferentially pumps out large hydrophobic, positively charged compounds such as vincristine, doxorubicin, actinomycin D, etoposide, etc.Citation3 Furthermore, P-gp is known to be overexpressed in many multidrug-resistant cancer cells and thus it plays an important role in the development of resistance to anti-cancer agents.Citation4
Breast cancer is a common cancer among women worldwide. Studies showed that estrogen receptor (ER) α-positive breast cancer MCF-7 cells in the presence of anti-estrogen drugs (e.g. tamoxifen) can increase epidermal growth factor receptor (EGFR) expression and the downstream signaling kinases (e.g., mitogen-activated protein kinase (MAPK)).Citation5 Extracellular signal–related kinase (ERK), a member of the MAPK pathway, plays an important role in cell proliferation, differentiation and cell migration.Citation6 Nevertheless, in breast cancer models, EGFR overexpression alone usually does not constitute tumorigenesis. However, the co-expression of EGFR with the non-receptor kinase c-Src significantly causes tumorigenesis, suggesting that EGFR and c-Src may functionally interact and contribute to the progression of breast cancer.Citation7
A protein tyrosine kinase (PTK) is an enzyme that modifies other proteins by transferring a phosphate group from ATP to a protein (i.e. phosphorylation) in cells. Tyrosine kinases have been identified as crucial factors in tumor progression. They promote cell division or inhibit apoptotic processes through the constitutive hyperactivation of downstream signaling cascades.Citation8 Furthermore, some tyrosine kinase inhibitors (TKIs) have been confirmed to be effective anticancer agents when administered alone or in combination with other conventional chemotherapeutic agents.Citation9 The interactions between TKIs and various ABC transporters have been extensively investigated in recent years. TKIs could selectively modulate the expressions or functions of ABC transporters via different mechanisms. One of the successful antitumor applications of TKIs is the development of BCR-Abl TKIs. It has been reported previously that imatinibCitation10 and nilotinibCitation11 both interact with specific ABC transporters and inhibit their transport activities significantly. Another type of potential antitumor TKIs is epidermal growth factor receptor (EGFR) TKIs, like gefitinib,Citation12 erlotinibCitation13 and lapatinib.Citation14 Vascular endothelial growth factor receptor (VEGFR) TKIs, such as vandetanibCitation15 and cediranib,Citation16 and the dual Abl-Src kinase inhibitors like bosutinibCitation17 together with multitargeted TKIs like sunitinibCitation18 have all been identified to significantly attenuate or reverse ABC transporters–mediated MDR in cancer cells. Therefore, it is possible that TKIs could be used as MDR reversal agents.
Dasatinib (BMS-354825), identified as a highly potent ATP-competitive inhibitor of both Src family kinases and Abl kinases,Citation19,20 was approved for imatinib resistant chronic myelogenous leukemia (CML) and (Ph+) acute lymphoblastic leukemia (ALL) treatment.Citation21,22 Though it has been reported previously that dasatinib is a dual BCRP/P-gp substrate,Citation23 no definite study has yet explored the role dasatinib plays in MDR. Dasatinib presents a moderate sensitivity to non-TNBC (“triple-negative” breast cancer) cell line MCF-7,Citation24 which may lead to a poor effect of dasatinib's cytotoxicity property on MCF-7 cells. In this study, we conducted a series of specific experiments to investigate the effect of dasatinib on the reversal of P-gp-mediated MDR in doxorubicin (DOX) – resistant MCF-7/Adr cells. We hope to expand the role of dasatinib in the treatment of breast cancer.
Materials and Methods
Reagents and antibodies
Dasatinib was purchased from Dalian Meilun Biotech Co., Ltd. (China). Verapamil hydrochloride was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (China). Both of them were dissolved in dimethyl sulphoxide (DMSO) and further diluted in culture medium with the final DMSO concentration <0.1%. DOX, obtained from Shenzhen Main Luck Pharmaceuticals Inc.. (China), was dissolved in saline and further diluted in culture medium. Mitogen-activated kinase kinase (MEK) inhibitor U0126 (Selleck Chemicals LLC Houston, TX, USA) was prepared in DMSO at 10 mM stock concentration. Monoclonal antibody against P-gp was from Abcam Company (UK). Phosphorylated extracellular signal–regulated kinase (P-ERK) and extracellular signal-regulated kinase (ERK) antibodies were from Cell Signaling Technology, Inc. (Beverly, MA, USA). Cleaved caspase-3 antibody and secondary antibodies were from Proteintech group (Chicago, USA). RPMI Medium 1640 and fetal bovine serum FBS were from GIBCO® (Grand Island, NY, USA). MTT was purchased from USB Corporation (Cleveland, OH, USA). All other chemicals were of analytical grade or purer.
Cell culture
Human breast carcinoma cell line MCF-7 and DOX-selected P-gp-overexpressing derivative MCF-7/Adr cells were purchased from Nanjing KeyGen Biotech Co. Ltd. (China), and cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acid solution, 100 U/ml penicillin and 0.1 mg/ml streptomycin. Vector-MDCK cell line was a generous gift from Professor Yuichi Sugiyama, Graduate School of Pharmaceutical Sciences, University of Tokyo (Japan). P-gp-MDCK transfectant cells were kindly provided by Professor Su Zeng, College of Pharmacy, Zhejiang University (China). Vector-MDCK and P-gp-MDCK cells were routinely maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acid solution, 100 U/ml penicillin and 0.1 mg/ml streptomycin. All cells were cultured at 37°C in a humidified atmosphere of 5% CO2. MCF-7/Adr cells were cultured in medium with 2 mg/mL DOX and were grown in DOX-free culture medium for more than 2 weeks before assay.
MTT cytotoxicity assay
Three–4,5-Dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide assay, known as MTT assay, was employed to assess the sensitivity of cells to drugs. Cells were collected and resuspended in a final concentration of 4 × 105 cells/ml, 100 μL aliquots were seeded in 96-well multiplates. After 24 h of culture, cells were exposed to a series of concentrations of DOX alone, or in combination with increasing concentrations of dasatinib or with 10 μM verapamil, and were cultured at 37˚C for 72 h. Then, MTT (5 mg/ml, 10 ml/well) was added to each well and cells were further incubated for 4 h at 37˚C, allowing the viable cells to reduce the yellow MTT into dark-blue formazan crystals, which were dissolved in 100 μl/well of SDS–isobutanol-HCl solution (10% SDS, 5% isobutanol, and 12 μM HCl) overnight at 37˚C. Optical density was measured at 570 nm with the microplate reader (Bio-Rad, San Diego, CA, USA). Six wells were used for each drug concentration and the experiment was replicated 3 times. The IC50 was calculated by Prism (Graphpad Software, La Jolla, CA, USA).Citation25 The fold-reversal factor of MDR was calculated by dividing the IC50 of DOX in the absence of dasatinib or verapamil by those obtained in the presence of dasatinib or verapamil. The resistance index was estimated by dividing the IC50 values of the MDR cells by that of the parental sensitive cells.
Doxorubicin accumulation assay
Cellular accumulation of doxorubicin was determined using the procedure originally notted by Ma MT et al.,Citation26 with minor modifications. Briefly, MDR cells were seeded in 6-well plates. After 24 h incubation of the cells with dasatinib or verapamil, DOX was added and incubated for 1 h at 37˚C. Cells were then washed with phosphate buffered saline (PBS), trypsinized and resuspended in medium at a concentration of 1×10Citation6 cells/ml. Intracellular fluorescence of DOX was determined by flow cytometry (FACScalibar, BD, USA) with excitation measured at 488 nm and emission measured at 575 nm.
Doxorubicin efflux assay
Doxorubicin efflux was assayed following a modified method previously described.Citation27 MCF-7/Adr cells were treated with 20 μM doxorubicin for 3h at 37°C, cells were then washed twice with ice-cold PBS, maintained at 37°C with DOX-absent culture media with or without 1.0 μM dasatinib for 0, 15, 30, 60 and 120 min respectively. Subsequent cells were then gathered and washed twice with ice-cold PBS. Finally, the cells were resuspended in ice-cold PBS buffer for flow cytometric analysis (FACScalibar, BD, USA), and the fluorescence intensity of DOX was determined.
TUNEL assay
After pre-incubating the MDR cells with dasatinib for 24 h, cells that grew on microscope slides were treated with DOX for additional 48 h based on IC50 value obtained from cell viability assay. Apoptotic cells were detected by measuring DNA fragmentation using the TdT-mediated dUTP nick-end labeling (TUNEL) method (In Situ Cell Death Detection Kit, POD, Roche Applied Science, Indianapolis, IN, USA) following the manufacturer's protocol. Briefly, cells were fixed with 4% paraformaldehyde for 25 min and washed with phosphate buffered saline (PBS) twice (5 min per time). Samples were treated with cyto-permeabilization solution 0.2% Triton X-100 and then washed again with PBS. A mixture of 5 μL TdT (terminal deoxynucleotidil transferase) enzyme and 45 μL fluorescein marked dUTP was added to each microscope slide. Finally, the slides were incubated for 1 h at 37˚C in a container to ensure proper moisture and darkness. Apoptotic cells were detected through a fluorescent microscope. Ten microscopic fields were randomly selected in each group to count the positive cells, and the extent of apoptosis was evaluated by the average number of positive cells (green spots) in the images.
Quantitative real-time PCR
Cellular P-gp mRNA expression was measured by Quantitative real-time PCR as previously described.Citation28 Total cellular RNA was extracted from cells using RNA isoPlus® Reagent Kit (Takara Biotechnology) according to the manufacturer's instructions. cDNA was generated from 1μg of total RNA using PrimeScript® RT Reagent Kit with gDNA Eraser (Takara Biotechnology) and was amplified using SYBR® Premix Ex Taq™ Kit (Takara Biotechnology) with β-actin as the normalization controls by ABI PRISM® 7500 Real-Time PCR System (Applied Biosystems). The fold change for MDR1 relative to the control was calculated using the comparative ΔΔCt method. Specific primers used for PCR amplification were as follows: MDR1 (Forward: 5′-GGAGCCTACTTGGTGGCACATAA-3′, reverse: 5′-TGGCATAGTCAGGAGCAAATGAAC-3′); β-actin (Forward:5′-ATTGAACACGGCATTGTCAC-3′, reverse: 5′-CATCGGAACCGCTCATTG-3′).
Western blot
As previously described,Citation29 cells were homogenized on ice in lysis buffer for Western blot analysis. Proteins from each cell lysate were mixed with an equal volume of 2 × Loading buffer (125 mM Tris–HCl, pH 6.8, 4% sodium dodecyl sulfate, 20% glycerol, 10% β-mercaptoethanol, and 0.05% bromophenol blue). Equal amounts of proteins were loaded and separated in sodium dodecyl sulfate–polyacrylamide gel by electrophoresis. Proteins were transferred onto PVDF membranes by electroblotting. Membranes were blocked with 5% milk or 5% BSA in Tris-buffered saline with 0.1% Tween 20 for 2 h at 37˚C, and then incubated with an appropriate dilution of antibodies overnight at 4˚C. Finally, blots were developed with corresponding horseradish peroxidase-conjugated anti-IgG antibodies and detected by an enhanced chemiluminescence (ECL) method using Bio-Spectrum Gel Imaging System (UVP, USA). Quantification of protein expression was analyzed through Gel-Pro Analyzer 3.0 software.
Statistical analysis
Data are expressed as means ± SD. All experiments were repeated at least 3 times and the differences were determined by using Student's t test. The value of P < 0.05 was considered to be statistically significant.
Results
Characterization of MCF-7 and MCF-7/Adr cells
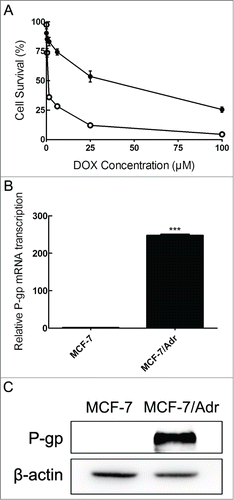
To validate the specific property that P-gp is overexpressed in MCF-7/Adr cell line, a comparison between MCF-7 and MCF-7/Adr cells about P-gp expression was provided. As shown in , MCF-7/Adr cells displayed lower cytotoxicity level of DOX than that of the parental MCF-7 cells. MCF-7/Adr revealed significantly higher levels of the mRNA and protein levels of P-gp in contrast with parental MCF-7 cells in . These results confirmed that P-gp is overexpressed in resistant MCF-7/Adr cells.
Figure 1. Characterization of MCF-7 and MCF-7/Adr cells. After treating cells with increasing concentrations of DOX in sensitive MCF-7 (○) and resistant MCF-7/Adr (•) cells for 48 h, cell viability was determined by MTT assay (A), mRNA expression of P-gp was tested by quantitative real-time PCR analysis (the control group is set at 1) (B) and P-gp protein level was detected by Western blot analysis (C). Data shown are means ± SD of triplicate determinations. ***P < 0.001, versus the control group.
Dasatinib potentiates the sensitivities of P-gp-overexpressing MCF-7/Adr cells to DOX
IC50 values of dasatinib determined by MTT assay for MCF-7 and MCF-7/Adr cells were 17.03 ± 1.93 and 40.10 ± 2.07 μM respectively. Based on the data tested, dasatinib was used at a maximum concentration of 1.0 μM – a concentration at which > 90% of the cells were viable to use in the next MDR reversal study. Dasatinib caused a dose-dependent decrease of the IC50 values of DOX compared to the significant decrease by P-gp inhibitor, verapamil in MCF-7/Adr cells. The fold-reversal of MDR was 1.57, 3.35 and 4.32 in the presence of 0.25, 0.5 and 1.0 μM dasatinib in MCF-7/Adr cells, respectively (). 1.0 μM dasatinib could also significantly decrease the IC50 values of DOX in stably transfected P-gp-MDCK cells in . Dasatinib barely altered the sensitization to DOX both in the drug-sensitive parental MCF-7 and MDCK cells. Therefore, the results suggested that dasatinib only significantly enhanced the sensitivity of P-gp-overexpressing cells to DOX.
Table 1. Effect of dasatinib on reversing P-gp-mediated MDR in drug selected cell line
Table 2. Effect of dasatinib on reversing P-gp-mediated MDR in transfected cell line
Dasatinib significantly increases the accumulation of DOX in P-gp-overexpressing MCF-7/Adr cells
To understand the mechanism by which dasatinib sensitizes the resistant cells to DOX, we examined the effect of dasatinib on the accumulation of DOX in P-gp-overexpressing MCF-7/Adr cells. The intracellular accumulation levels of DOX in sensitive MCF-7 cells were higher than that of resistant MCF-7/Adr cells (). Intracellular accumulation of DOX in the absence of dasatinib was low. However, dasatinib significantly increased the intracellular accumulation of DOX in a concentration-dependent manner, compared to the positive control group of verapamil. Dasatinib at 0.25, 0.5 and 1.0 μM concentrations increased the intracellular accumulation of DOX by 1.06-, 1.23- and 1.40-fold in MCF-7/Adr cells, respectively. It had little impact on the intracellular accumulation of DOX in the parental sensitive MCF-7 cells. In summary, these results suggested that dasatinib significantly inhibited P-gp-mediated transport in resistant MCF-7/Adr cells.
Figure 2. Effects of dasatinib on the accumulation and efflux of DOX. (A) Cells were pre-incubated with or without dasatinib or verapamil for 24 h at 37°C and then incubated with 20 μM DOX for another 1h at 37°C. The results are presented as fold change in fluorescence intensity relative to untreated control resistant cells. (B) After a preincubation of 20 μM DOX for 3 h, time course of DOX efflux was measured in MCF-7/Adr cells with the presence (•) or absence (○) of 1.0 μM dasatinib. The accumulation of DOX was measured by flow cytometric analysis. Data shown are means ± SD of triplicate determinations. *P < 0.05 and **P < 0.01 vs. the control group.
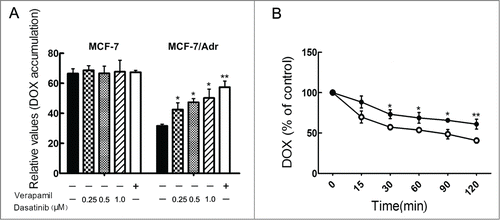
Dasatinib inhibits the efflux of DOX in P-gp-overexpressing MCF-7/Adr cells
As dasatinib increased the intracellular accumulation of DOX in MCF-7/Adr cells, we determined that the increased accumulation was due to the inhibition of its efflux. The course of DOX efflux within 2 h after accumulation is shown in Fig. 2B. Dasatinib inhibited DOX efflux of P-gp in MCF-7/Adr cells while it made little difference on DOX efflux in sensitive MCF-7 cells. For instance, at 120 min, 36.9% of accumulated DOX was pumped out of MCF-7/Adr cells in the presence of 1.0 μM dasatinib while 55.4% of accumulated DOX in MCF-7/Adr cells was effluxed in the absence of dasatinib (P < 0.05). The results indicated that dasatinib could effectively inhibit DOX efflux of P-gp.
Dasatinib significantly enhances DOX-induced apoptosis in resistant MCF-7/Adr cells
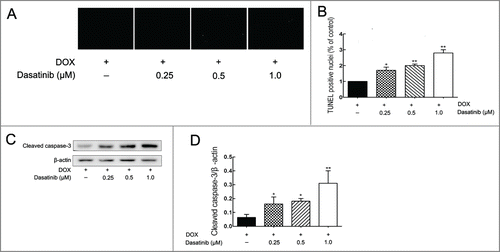
TUNEL assay showed that dasatinib produced a dose-dependent increase in the number of apoptotic MCF-7/Adr cells induced by DOX in . This may have resulted from an enhanced activation of caspase-3 in . Results confirmed that dasatinib could enhance DOX-induced apoptosis in resistant MCF-7/Adr cells.
Figure 3. Effect of dasatinib on DOX-induced cell apoptosis. (A) After pre-incubating the cells with dasatinib for 24 h at 37°C, cells grown on coverslips were treated with the 20 μM DOX for another 48 h, and apoptosis was analyzed by TUNEL assay as described in Materials and Methods. (B) Quantification of TUNEL fluorescent images. After pre-incubating the cells with dasatinib for 24 h, cleaved caspase-3 expression was performed (C) with quantification (D). Each column shows the mean ± SD of 3 independent experiments, performed in triplicate. *P < 0.05 and **P < 0.01 versus the control group.
Dasatinib significantly alters the mRNA and protein levels of P-gp
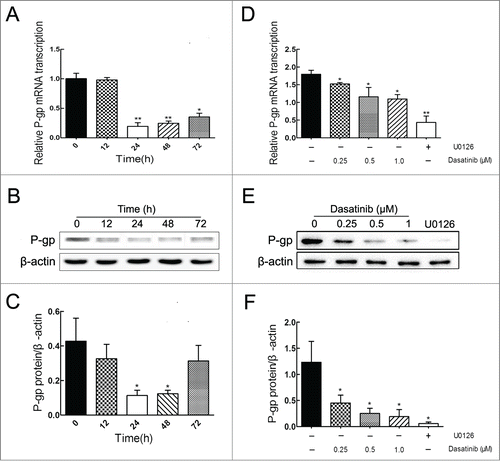
To confirm that the reversal of P-gp-mediated MDR is attributed to a reduction of P-gp expression, we determined the effects of dasatinib on the expression levels of mRNA and protein levels of P-gp in resistant MCF-7/Adr cells. Results showed that dasatinib did significantly downregulate the expressions of mRNA and protein levels of P-gp both in time-dependent (before 24 h) () and dose-dependent () manners in MCF-7/Adr cells. These data suggested that the reversal effect of dasatinib on P-gp-mediated MDR was ascribed to the downregulation of P-gp expression in MCF-7/Adr cells.
Figure 4. Effects of dasatinib on the mRNA and protein expressions of P-gp in resistant MCF-7/Adr cells. (A, B) Cells were treated with 1.0 μM dasatinib at the indicated time of 0, 12, 24, 48, 72 h, and dasatinib downregulated P-gp in a time-dependent manner before 24 h. (D, E) Cells were treated with 10 μM U0126 and dasatinib at the indicated concentrations of 0, 0.25, 0.5, 1.0 μM for 24 h, and dasatinib downregulated P-gp in a dose-dependent manner. (C, F) The protein expression of P-gp was normalized to β-actin. Each column shows the mean ± SD of 3 independent experiments, performed in triplicate. *P < 0.05 and **P < 0.01, vs. the control group.
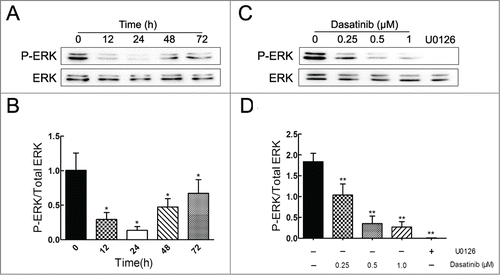
Dasatinib significantly blocks the phosphorylation of ERK
To further investigate the potential mechanism involved in the downregulation of P-gp, we determined the effects of dasatinib on the levels of total and phosphorylated forms of ERK in MCF-7/Adr cells. Results showed that dasatinib remarkably decreased the phosphorylated form of ERK both in time-dependent (before 24 h) () and dose-dependent () manners compared to the positive group of MEK inhibitor U0126, whereas it had no effect on the total form of ERK. In view of U0126's significant downregulation of P-gp in , the result indicated that the reversal effect of dasatinib on P-gp-mediated MDR might be associated with the inhibition of ERK pathway in MCF-7/Adr cells.
Figure 5. Effect of dasatinib on the expression of phospho-ERK. P-ERK expression was assayed with the treatment of 1.0 μM dasatinib at the indicated time of 0, 12, 24, 48, 72 h (A) and at the indicated concentrations of 0, 0.25, 0.5, 1.0 μM for 24 h (C). U0126 acts as the positive control group. (B, D) The quantification of P-ERK expression was normalized to the total content of ERK. Each column shows the mean ± SD of 3 independent experiments, performed in triplicate. *P < 0.05 and **P < 0.01, versus the control group.
Discussion
Breast cancer is now the second leading cause of cancer among females, next only to lung cancer.Citation30 Despite significant advances in the diagnosis and treatment of breast cancer, metastatic breast cancer is still incurable. Therefore, identification of the key molecular alterations that induce proliferation or drug resistance in breast cancer cells are needed to help discover potential therapeutic targets. Researchers have long understood some of the key molecular drivers of breast cancer, using them as prognostic factors and/or predictive markers for specific therapies such as estrogen receptor (ER), progesterone receptor (PgR), and HER2 and so forth. According to Peyton Rous,Citation31 Src is the earliest known oncogene identified as the transforming agent in chicken sarcomas. Previous studies reported that Src kinase activity was 4- to 20- fold higher than normal tissues in human mammary carcinomas.Citation32-34 Anomalous activation or amplification of SRC and SRC-family kinases (SFK) has been identified to play an important role in proliferation, migration, and invasion of breast cancer cell lines.Citation35,36 In addition, Abl kinases, one of whose activated forms is BCR-Abl, plays a functional role in breast cancer.Citation37,38 Thus, both Src and Abl kinases represent effective molecular targets for the development and progression of breast cancer.
Dasatinib is a potent dual Src/Abl kinase inhibitor, implying that dasatinib may be closely associated with the evolvement of breast cancer. As previously reported, dasatinib have an effect on TNBC which is generally the basal-type breast cancer.Citation24,39 Furthermore, according to Richard S. Finn et al.,Citation35 that basal-type and epithelial-tomesenchymal transition (post-EMT) breast cancer cell lines are more sensitive than luminal-type breast cancer cells to growth inhibition by dasatinib. Thus, based on the low sensitivity of luminal breast cancer cell lines to dasatinib, we on the other side aimed to explore the effect of dasatinib on MDR in one kind of luminal breast cancer cell lines MCF-7.
In the present study, MCF-7/Adr cells were verified to be DOX-resistant (), via mechanisms that include the overexpression of mRNA and protein levels of P-gp (). Studies also showed that reversal concentrations of dasatinib remarkably potentiated the cytotoxicity of DOX in P-gp-overexpressing MCF-7/Adr and P-gp-transfected MDCK cells, but did not sensitize the parental sensitive MCF-7 or MDCK cells to DOX as shown in . Dasatinib significantly increased the accumulation () and inhibited the efflux () of DOX in resistant MCF-7/Adr cells.
P-gp, apart from its role as an efflux pump, can also regulate programmed cell death induced by antineoplastic agents like DOX,Citation40 UV irradiation and ligation of the cell surface death receptors Fas. Generally, these diverse apoptotic stimuli initiate cell death by activation of caspases like caspase-3.Citation41-43 However, it has been reported that functional P-gp could inhibit the activation of caspase-3, which could be reversed by inhibiting P-gp.Citation42 Therefore, dasatinib could increase cell apoptosis in a dose-dependent manner () with an enhanced activation of caspase-3 (). These findings suggested that dasatinib could enhance DOX-induced apoptosis in MDR cells.
For initial investigation, we suggested that the MDR reversal effect of dasatinib is due to either the inhibition of P-gp efflux or the downregulation of P-gp expression, or both. Our results showed that dasatinib downregulated the mRNA and protein levels of P-gp both in time-dependent (before 24 h) () and dose-dependent manners () in resistant MCF-7/Adr cells. On the other hand, it has been reported that for dasatinib, the ATPase assays did not show a measurable effect on the P-gp ATPase activity up to 1 μM concentration.Citation17 Therefore, it could be suggested that dasatinib reverses P-gp-mediated MDR by downregulating P-gp expression as opposed to the direct inhibition of P-gp efflux.
Previous studies also demonstrated that dasatinib inhibited cell activation and even induced apoptosis by blocking downstream signaling pathways like phosphorylation of ERK.Citation44, 45 And the mechanism of anti-MDR may be associated with the inhibition of ERK pathway.Citation46-48 In this study, we analyzed the possible association between ERK pathway and the MDR reversal effect of dasatinib. Results revealed that reversal concentrations of dasatinib significantly blocked the phosphorylation of ERK both in time-dependent (before 24 h) () and dose-dependent () manners in MCF-7/Adr cells. U0126's significant downregulation of P-gp in indicated that the inhibition of ERK is involved in the reversal of P-gp-mediated MDR by dasatinib.
In conclusion, this study provides an evidence that dasatinib could significantly enhance the efficacy of chemotherapeutic drug in P-gp-overexpressing MDR cells. Furthermore, the reversal of MDR by dasatinib was ascribed to the downregulation of both the mRNA and protein levels of P-gp as opposed to the direct inhibition of P-gp efflux. The downregulation of P-gp might partly depend on the inhibition of ERK pathway. Confirmation of MDR reversal by dasatinib may support the potential benefits of combining dasatinib with other conventional antineoplastic drugs in overcoming drug resistance in cancer chemotherapy.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
987062_Supplementary_Materials.zip
Download Zip (1.3 MB)Acknowledgments
We wish to express our heartfelt gratitude to Dr. Yuichi Sugiyama (Graduate School of Pharmaceutical Sciences, University of Tokyo, Tokyo, Japan) for providing vector – MDCK cells, likewise, we also wish to express our sincere gratitude to Professor Zeng Su (College of Pharmacy, Zhejiang University, China) for providing stable transfected ABCB1-MDCK cells.
Funding
This article was supported by a grant from the National Natural Science Foundation of China (No. 81273580, 81072694).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Litman T, Druley TE, Stein WD, Bates SE. From MDR to MXR: new understanding of multidrug resistance systems, their properties and clinical significance. Cell Mol Life Sci 2001; 58: 931-59; PMID:11497241; http://dx.doi.org/10.1007/PL00000912
- Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 1976; 455: 152-62; PMID:990323; http://dx.doi.org/10.1016/0005-2736(76)90160-7
- Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene 2003; 22: 7468-85; PMID:14576852; http://dx.doi.org/10.1038/sj.onc.1206948
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2002; 2: 48-58; PMID:11902585; http://dx.doi.org/10.1038/nrc706
- Ghayad SE, Vendrell JA, Ben Larbi S, Dumontet C, Bieche I, Cohen PA. Endocrine resistance associated with activated ErbB system in breast cancer cells is reversed by inhibiting MAPK or PI3K/Akt signaling pathways. Int J Cancer 2010; 126: 545-62; PMID:19609946; http://dx.doi.org/10.1002/ijc.24750
- McCawley LJ, Li S, Wattenberg EV, Hudson LG. Sustained activation of the mitogen-activated protein kinase pathway. A mechanism underlying receptor tyrosine kinase specificity for matrix metalloproteinase-9 induction and cell migration. J Biol Chem 1999; 274: 4347-53; PMID:9933637; http://dx.doi.org/10.1074/jbc.274.7.4347
- Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl Acad Sci U S A 1999; 96: 1415-20; PMID:9990038; http://dx.doi.org/10.1073/pnas.96.4.1415
- Sawyers C. Targeted cancer therapy. Nature 2004; 432: 294-7; PMID:15549090; http://dx.doi.org/10.1038/nature03095
- Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther 2005; 315: 971-9; PMID:16002463; http://dx.doi.org/10.1124/jpet.105.084145
- Houghton PJ, Germain GS, Harwood FC, Schuetz JD, Stewart CF, Buchdunger E, Traxler P. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res 2004; 64: 2333-7; PMID:15059881; http://dx.doi.org/10.1158/0008-5472.CAN-03-3344
- Tiwari AK, Sodani K, Wang SR, Kuang YH, Ashby CR, Jr., Chen X, Chen ZS. Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem Pharmacol 2009; 78: 153-61; PMID:19427995; http://dx.doi.org/10.1016/j.bcp.2009.04.002
- Kitazaki T, Oka M, Nakamura Y, Tsurutani J, Doi S, Yasunaga M, Yabuuchi H, Soda H, Kohno S. Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer 2005; 49: 337-43; PMID:15955594; http://dx.doi.org/10.1016/j.lungcan.2005.03.035
- Shi Z, Peng XX, Kim IW, Shukla S, Si QS, Robey RW, Bates SE, Shen T, Ashby CR Jr, Fu LW, et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res 2007; 67: 11012-20; PMID:18006847; http://dx.doi.org/10.1158/0008-5472.CAN-07-2686
- Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG, Ashby CR Jr, Huang Y, Robey RW, Liang YJ, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res 2008; 68: 7905-14; PMID:18829547; http://dx.doi.org/10.1158/0008-5472.CAN-08-0499
- Zheng LS, Wang F, Li YH, Zhang X, Chen LM, Liang YJ, Dai CL, Yan YY, Tao LY, Mi YJ, et al. Vandetanib (Zactima, ZD6474) antagonizes ABCC1- and ABCG2-mediated multidrug resistance by inhibition of their transport function. PLoS One 2009; 4: e5172
- Tao LY, Liang YJ, Wang F, Chen LM, Yan YY, Dai CL, Fu LW. Cediranib (recentin, AZD2171) reverses ABCB1- and ABCC1-mediated multidrug resistance by inhibition of their transport function. Cancer Chemother Pharmacol 2009; 64: 961-9; PMID:19255759; http://dx.doi.org/10.1007/s00280-009-0949-1
- Hegedus C, Ozvegy-Laczka C, Apati A, Magocsi M, Nemet K, Orfi L, Kéri G, Katona M, Takáts Z, Váradi A, et al. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: implications for altered anti-cancer effects and pharmacological properties. Br J Pharmacol 2009; 158: 1153-64; PMID:19785662; http://dx.doi.org/10.1111/j.1476-5381.2009.00383.x
- Dai CL, Liang YJ, Wang YS, Tiwari AK, Yan YY, Wang F, Chen ZS, Tong XZ, Fu LW. Sensitization of ABCG2-overexpressing cells to conventional chemotherapeutic agent by sunitinib was associated with inhibiting the function of ABCG2. Cancer Lett 2009; 279: 74-83; PMID:19232821; http://dx.doi.org/10.1016/j.canlet.2009.01.027
- Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, Castaneda S, Cornelius LA, Das J, Doweyko AM, et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem 2004; 47: 6658-61; PMID:15615512; http://dx.doi.org/10.1021/jm049486a
- Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004; 305: 399-401; PMID:15256671; http://dx.doi.org/10.1126/science.1099480
- Steinberg M. Dasatinib: a tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia and philadelphia chromosome-positive acute lymphoblastic leukemia. Clin Ther 2007; 29: 2289-308; PMID:18158072; http://dx.doi.org/10.1016/j.clinthera.2007.11.005
- Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, Cortes J, O'Brien S, Nicaise C, Bleickardt E, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med 2006; 354: 2531-41; PMID:16775234; http://dx.doi.org/10.1056/NEJMoa055229
- Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther 2009; 330: 956-63; PMID:19491323; http://dx.doi.org/10.1124/jpet.109.154781
- Finn RS, Bengala C, Ibrahim N, Roche H, Sparano J, Strauss LC, Fairchild J, Sy O, Goldstein LJ. Dasatinib as a single agent in triple-negative breast cancer: results of an open-label phase 2 study. Clin Cancer Res 2011; 17: 6905-13; PMID:22028489; http://dx.doi.org/10.1158/1078-0432.CCR-11-0288
- Zhu Y, Meng Q, Wang C, Liu Q, Huo X, Zhang A, Sun P, Sun H, Li H, Liu K. Methotrexate-bestatin interaction: Involvement of P-glycoprotein and organic anion transporters in rats. Int J Pharm 2014; 465: 368-77; PMID:24530518; http://dx.doi.org/10.1016/j.ijpharm.2014.02.020
- Ma MT, He M, Wang Y, Jiao XY, Zhao L, Bai XF, Yu ZJ, Wu HZ, Sun ML, Song ZG, et al. MiR-487a resensitizes mitoxantrone (MX)-resistant breast cancer cells (MCF-7/MX) to MX by targeting breast cancer resistance protein (BCRP/ABCG2). Cancer Lett 2013; 339: 107-15; PMID:23879965; http://dx.doi.org/10.1016/j.canlet.2013.07.016
- Zhou WJ, Zhang X, Cheng C, Wang F, Wang XK, Liang YJ, To KK, Zhou W, Huang HB, Fu LW. Crizotinib (PF-02341066) reverses multidrug resistance in cancer cells by inhibiting the function of P-glycoprotein. Br J Pharmacol 2012; 166: 1669-83; PMID:22233293; http://dx.doi.org/10.1111/j.1476-5381.2012.01849.x
- Huo X, Liu Q, Wang C, Meng Q, Sun H, Peng J, Ma X, Liu K. Enhancement effect of P-gp inhibitors on the intestinal absorption and antiproliferative activity of bestatin. Eur J Pharm Sci 2013; 50: 420-8; PMID:23981338; http://dx.doi.org/10.1016/j.ejps.2013.08.010
- Wang L, Meng Q, Wang C, Liu Q, Peng J, Huo X, Sun H, Ma X, Liu K. Dioscin restores the activity of the anticancer agent adriamycin in multidrug-resistant human leukemia K562/adriamycin cells by down-regulating MDR1 via a mechanism involving NF-kappaB signaling inhibition. J Nat Prod 2013; 76: 909-14; PMID:23621869; http://dx.doi.org/10.1021/np400071c
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin 2008; 58:71-96; PMID:18287387; http://dx.doi.org/10.3322/CA.2007.0010
- Rous PA. Sarcoma of the Fowl Transmissible by an Agent Separable from the Tumor Cells. J Exp Med 1911; 13: 397-411; PMID:19867421; http://dx.doi.org/10.1084/jem.13.4.397
- Ottenhoff-Kalff AE, Rijksen G, van Beurden EA, Hennipman A, Michels AA, Staal GE. Characterization of protein tyrosine kinases from human breast cancer: involvement of the c-src oncogene product. Cancer Res 1992; 52: 4773-8; PMID:1380891
- Jacobs C, Rubsamen H. Expression of pp60c-src protein kinase in adult and fetal human tissue: high activities in some sarcomas and mammary carcinomas. Cancer Res 1983; 43: 1696-702; PMID:6403227
- Muthuswamy SK, Siegel PM, Dankort DL, Webster MA, Muller WJ. Mammary tumors expressing the neu proto-oncogene possess elevated c-Src tyrosine kinase activity. Mol Cell Biol 1994; 14: 735-43; PMID:7903421
- Finn RS, Dering J, Ginther C, Wilson CA, Glaspy P, Tchekmedyian N, Slamon DJ. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/"triple-negative" breast cancer cell lines growing in vitro. Breast Cancer Res Treat 2007; 105: 319-26; PMID:17268817; http://dx.doi.org/10.1007/s10549-006-9463-x
- Choi YL, Bocanegra M, Kwon MJ, Shin YK, Nam SJ, Yang JH, Kao J, Godwin AK, Pollack JR. LYN is a mediator of epithelial-mesenchymal transition and a target of dasatinib in breast cancer. Cancer Res 2010; 70: 2296-306; PMID:20215510; http://dx.doi.org/10.1158/0008-5472.CAN-09-3141
- Srinivasan D, Plattner R. Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res 2006; 66: 5648-55; PMID:16740702; http://dx.doi.org/10.1158/0008-5472.CAN-06-0734
- Srinivasan D, Sims JT, Plattner R. Aggressive breast cancer cells are dependent on activated Abl kinases for proliferation, anchorage-independent growth and survival. Oncogene 2008; 27: 1095-105; PMID:17700528; http://dx.doi.org/10.1038/sj.onc.1210714
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011; 121: 2750-67; PMID:21633166; http://dx.doi.org/10.1172/JCI45014
- Zhao N, Wang R, Zhou L, Zhu Y, Gong J, Zhuang SM. MicroRNA-26b suppresses the NF-kappaB signaling and enhances the chemosensitivity of hepatocellular carcinoma cells by targeting TAK1 and TAB3. Mol Cancer 2014; 13: 35; PMID:24565101; http://dx.doi.org/10.1186/1476-4598-13-35
- Smyth MJ, Krasovskis E, Sutton VR, Johnstone RW. The drug efflux protein, P-glycoprotein, additionally protects drug-resistant tumor cells from multiple forms of caspase-dependent apoptosis. Proc Natl Acad Sci U S A 1998; 95: 7024-9; PMID:9618532; http://dx.doi.org/10.1073/pnas.95.12.7024
- Johnstone RW, Cretney E, Smyth MJ. P-glycoprotein protects leukemia cells against caspase-dependent, but not caspase-independent, cell death. Blood 1999; 93: 1075-85; PMID:9920858
- Robinson LJ, Roberts WK, Ling TT, Lamming D, Sternberg SS, Roepe PD. Human MDR 1 protein overexpression delays the apoptotic cascade in Chinese hamster ovary fibroblasts. Biochemistry 1997; 36: 11169-78; PMID:9287159; http://dx.doi.org/10.1021/bi9627830
- Salih J, Hilpert J, Placke T, Grunebach F, Steinle A, Salih HR, Krusch M. The BCR/ABL-inhibitors imatinib, nilotinib and dasatinib differentially affect NK cell reactivity. Int J Cancer 2010; 127: 2119-28; PMID:20143399; http://dx.doi.org/10.1002/ijc.25233
- Dai Y, Chen S, Shah R, Pei XY, Wang L, Almenara JA, Kramer LB, Dent P, Grant S. Disruption of Src function potentiates Chk1-inhibitor-induced apoptosis in human multiple myeloma cells in vitro and in vivo. Blood 2011; 117: 1947-57; PMID:21148814; http://dx.doi.org/10.1182/blood-2010-06-291146
- Wei N, Liu GT, Chen XG, Liu Q, Wang FP, Sun H. H1, a derivative of Tetrandrine, exerts anti-MDR activity by initiating intrinsic apoptosis pathway and inhibiting the activation of Erk1/2 and Akt1/2. Biochem Pharmacol 2011; 82: 1593-603; PMID:21864508; http://dx.doi.org/10.1016/j.bcp.2011.08.012
- Oh SY, Song JH, Gil JE, Kim JH, Yeom YI, Moon EY. ERK activation by thymosin-beta-4 (TB4) overexpression induces paclitaxel-resistance. Exp Cell Res 2006; 312: 1651-7; PMID:16515784; http://dx.doi.org/10.1016/j.yexcr.2006.01.030
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 2007; 1773: 1263-84; PMID:17126425; http://dx.doi.org/ 10.1016/j.bbamcr.2006.10.001