
Abstract
Site-specific hypermethylation of tumor suppressor genes accompanied by genome-wide hypomethylation are epigenetic hallmarks of malignancy. However, the molecular mechanisms that drive these linked changes in DNA methylation remain obscure. DNA methyltransferase 1 (DNMT1), the principle enzyme responsible for maintaining methylation patterns is commonly dysregulated in tumors. Replication foci targeting sequence (RFTS) is an N-terminal domain of DNMT1 that inhibits DNA-binding and catalytic activity, suggesting that RFTS deletion would result in a gain of DNMT1 function. However, a substantial body of data suggested that RFTS is required for DNMT1 activity. Here, we demonstrate that deletion of RFTS alters DNMT1-dependent DNA methylation during malignant transformation. Compared to full-length DNMT1, ectopic expression of hyperactive DNMT1-ΔRFTS caused greater malignant transformation and enhanced promoter methylation with condensed chromatin structure that silenced DAPK and DUOX1 expression. Simultaneously, deletion of RFTS impaired DNMT1 chromatin association with pericentromeric Satellite 2 (SAT2) repeat sequences and produced DNA demethylation at SAT2 repeats and globally. To our knowledge, RFTS-deleted DNMT1 is the first single factor that can reprogram focal hypermethylation and global hypomethylation in parallel during malignant transformation. Our evidence suggests that the RFTS domain of DNMT1 is a target responsible for epigenetic changes in cancer.
Abbreviations
| TSGs | = | tumor suppressor genes |
| DNMT1 | = | DNA methyltransferase 1 |
| RFTS | = | replication foci targeting sequence |
| HELP | = | HpaII tiny fragment enrichment by ligation-mediated PCR |
| KEGG | = | Kyoto Encyclopedia of Genes and Genomes |
| COSMIC | = | catalog of somatic mutation in cancer |
Introduction
Methylation of cytosine bases in CpG dinucleotides is an epigenetic modification in mammals involved in development, imprinting and X chromosome inactivation, which becomes dysregulated in carcinogenesis.Citation1 Increased DNA methylation in promoter regions is highly associated with transcriptional silencing and hypermethylation-mediated silencing of tumor suppressor genes (TSGs) is a critical epigenetic driver of cancer.Citation2,3 However, in addition to site-specific hypermethylation, a reduction of total cytosine methylation of the genome, targeted in part to repetitive elements, is also common in malignancies.Citation4-6 Global hypomethylation may be a driver of cancer by promoting genomic instability and oncogene activation. Although both hypermethylation and hypomethylation are found in cancer, the molecular mechanisms that link these alterations are not clear.Citation7
DNA methyltransferase 1 (DNMT1) is the enzyme most responsible for the maintenance of DNA methylation patterns during DNA replication. In addition to maintenance methylation activity, DNMT1 can perform de novo DNA methylation as do DNMT3A and DNMT3B.Citation8-10 Dysregulated DNMT1 is capable of promoting hypermethylation or hypomethylation and, indeed, DNMT1 has been reported to be up-regulated or down-regulated in different types of cancer.Citation11–14 The methyltransferase activity of DNMT1 has been considered the critical effector in reprogramming cancer methylation patterns. However, enforced overexpression of DNMT1 produces hypermethylation of TSGs and increases methylation of the whole genome.Citation15-17 Conversely, deficient DNMT1 causes global hypomethylation without site-specific hypermethylation.Citation18,19 Thus, changing the expression of DNMT1 alone is insufficient to recapitulate the linked and vectorially opposite epigenetic alterations observed in cancer.
DNMT1 consists of a series of globular domains N-terminal to the catalytic domain, which are implicated in functions essential for catalytic activity, protein association and target specificity. By interacting with different proteins, DNMT1 may be enriched in or dissociated from specific genomic lociCitation20-26 with the distribution of DNMT1 determining the methylation status of specific target sites. For example, the oncogenic PML-RAR fusion protein recruits DNMT1 to the RARβ2 promoter to stimulate methylation,Citation20 whereas disruption of the DNMT1-proliferating cell nuclear antigen (PCNA) interaction results in hypomethylation of cellular DNA.Citation24,25 Seemingly, both enzyme activity and chromatin occupancy of DNMT1 are important for DNMT1-dependent changes in DNA methylation observed during tumorigenesis.
The replication foci targeting sequence (RFTS) was defined as an N-terminal domain required for associating DNMT1 with replication foci.Citation27 Multiple lines of evidence suggested that RFTS is a positively acting domain required for DNMT1 function.Citation27-30 Surprisingly, recombinant forms of DNMT1 including the RFTS domain bind DNA poorly, while specific deletion of RFTS activates DNA binding.Citation31 Moreover, using a fluorigenic methylation assay, we showed that deletion of RFTS leads to a 640-fold increase in methylation activity due to relief of DNA-competitive inhibition by RFTS. Further biochemical and structural experiments support RFTS as a DNA-competitive endogenous inhibitor of DNMT1 that must be removed from the DNA active site for DNMT1 activity.Citation32–36 In contrast to our biochemical insights into negative regulation by RFTS, CXXC was proposed as a nonmethylated DNA-binding inhibitor of DNMT1 activity.Citation37 Human genetics has the potential to help resolve DNMT1 domain function because an autosomal dominant hereditary sensory and autonomic neuropathy (HSAN1E) maps to the RFTS domain.Citation38 This mutation is associated with genomic hypomethylation and promoter hypermethylation.Citation38
In this study, we investigate the functional impact of 2 DNMT1 regulatory domains, RFTS and CXXC. Here we show that expression of full-length and various deletion mutants resulted in degrees of promoter hypermethylation, chromatin condensation, and transcriptional repression. Ectopic expression of DNMT1-ΔRFTS—at a level equivalent to endogenous DNMT1—led to the most malignant phenotype, which required the presence of the CXXC domain for oncogenic activity. Surprisingly, DNMT1-ΔRFTS cells further showed a global hypomethylation phenotype reminiscent of the malignant state. These data suggest that tumor-specific modulation of RFTS function may mediate the commonly observed linkage of focal hypermethylation and global hypomethylation.
Results
Deletion of RFTS enhances the oncogenic activity of DNMT1
To dissect the functional role of 2 regulatory domains in human DNMT1, we established stable cell lines in non-malignant human bronchial epithelial cells (HBEC3)Citation39 that expressed full-length DNMT1 (DNMT1 cells), RFTS-deleted DNMT1 (DNMT1-ΔRFTS cells), CXXC-deleted DNMT1 (DNMT1-ΔCXXC cells), RFTS/CXXC double-deleted DNMT1 (DNMT1-ΔR/C cells), or vector alone (vector cells). mRNA and protein validation showed that each DNMT1 construct was expressed at levels equivalent to that of endogenous DNMT1 in HBEC3 (). Because DNMT1 has protooncogenic properties,Citation15,16,40 we evaluated transformation markers of the resulting cell lines. There was no significant proliferation difference between vector cells and DNMT1-expressing cells, with or without supplementation of epidermal growth factor (EGF) (Fig. S1A). We then examined whether DNMT1 overexpression could alter invasive activity, a hallmark of cancer metastasis (Fig. S1B). Although invasion was slightly enhanced in all DNMT1-expressing cells, only DNMT1-ΔRFTS and DNMT1-ΔR/C cells showed a significant difference compared to vector cells, suggesting that deletion of the RFTS domain provides an invasive advantage.
Figure 1. Deletion of RFTS enhances the oncogenic activity of DNMT1. (A) HBEC3 stable cell lines were established to express full-length and DNMT1 deletion forms near endogenous DNMT1 levels. The levels of DNMT1 were determined by RT-qPCR (left) and western blotting (right). Data were normalized to vector cells. (B) Adherent colony formation. (C) Soft-agar colony formation. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, indicates no significant difference in comparison to vector cells.
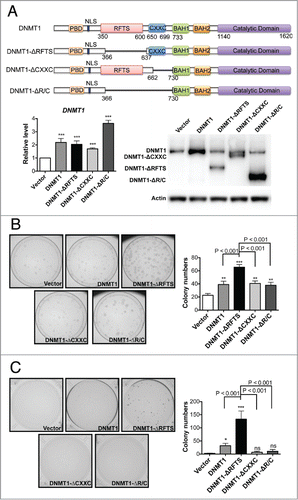
To further analyze the oncogenic properties of these cells, we examined anchorage-dependent growth (). Overexpression of all forms of DNMT1 increased the number of colonies observed. However, whereas DNMT1 cells produced 2-fold more colonies, DNMT1-ΔRFTS cells produced 3-fold more colonies. We further tested the anchorage-independent colony-forming activity of these cells by soft-agar colony formation (). Because HBEC3 is a non-malignant cell line, vector cells produced fewer than 5 colonies per dish. However, DNMT1 cells produced 30 colonies and DNMT1-ΔRFTS cells produced 130 colonies in soft agar. Interestingly, though DNMT1-ΔRFTS cells elevated soft-agar colony-forming activity, removing the CXXC domain in DNMT1-ΔR/C cells completely abolished the effect. These data indicate that the CXXC domain is essential for the oncogenic activity of DNMT1-ΔRFTS. Thus, DNMT1 acts as weak oncoprotein, whose transforming activity is enhanced by deletion of the inhibitory RFTS domain.
Promoter hypermethylation and transcriptional silencing of DAPK and DUOX1 is driven by DNMT1
Several TSGs are subject to methylation-associated silencing in lung cancer (Table S1). To test whether introduction of ectopic DNMT1 alleles is sufficient to alter DNA methylation of these genes, we examined the methylation status of promoter-associated CpG islands and transcript levels for each of 24 TSGs. Of these genes, DAPK Citation41,42 and DUOX1Citation43 were consistently hypermethylated and silenced by all DNMT1 alleles. By performing methylated DNA immunoprecipitation (MeDIP), we found a 2-fold increase in methylation of the DAPK promoter in all DNMT1-expressing cells (). Bisulfite sequencing indicated that there were few methylated CpGs in vector cells (1.6%) (). DNMT1 and DNMT1-ΔRFTS overexpression upregulated methylation more than 10-fold to 17.6% and 24.6%, respectively. Although there was no apparent difference between DNMT1 and DNMT1-ΔRFTS from MeDIP analysis, bisulfite sequencing showed that DNMT1-ΔRFTS expression drove greater methylation of the DAPK promoter than did wild-type DNMT1. Furthermore, we analyzed the association between DNMT1 and the DAPK promoter region by DNMT1 chromatin immunoprecipitation (ChIP) (). We found increased chromatin occupancy of DNMT1 at the DAPK promoter in all DNMT1-expressing cells.
Figure 2. DNMT1-ΔRFTS promotes increased methylation and silencing of the DAPK and DUOX1 genes. (A) The methylation levels of DAPK (left) and DUOX1 (right) promoter-associated CpG islands were analyzed by qPCR. Methylated DNA was analyzed using the MethylMiner kit and amplified with specific primers. (B) Bisulfite sequencing results for DAPK (left) and DUOX1 (right) promoters. White squares represent unmethylated cytosines and black squares represent methylated cytosines in CpG sites. The percentage of methylated CpG dinucleotides from 8 independent clones is indicated. (C) DNMT1 chromatin occupancy was analyzed using DNMT1 ChIP and qPCR. (D) mRNA levels of DAPK (left) and DUOX1 (right) were analyzed by RT-qPCR and normalized to vector cells. *, p < 0.05; **, p < 0.01; ***, p < 0.001 in comparison to vector cells.
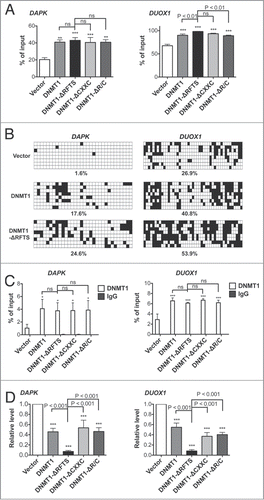
The transcript levels of DAPK were consistent with its increased promoter methylation. Compared to the vector control, expression of DAPK was decreased in all DNMT1-expressing cells, among which DNMT1-ΔRFTS cells showed the greatest repressive effect (). A similar correlation in DNA methylation, DNMT1 chromatin occupancy and mRNA expression were also found in DUOX1 in all DNMT1-expressing cells (). In agreement with the transformation result, DNMT1-ΔRFTS overexpression resulted in the greatest effect on hypermethylation-mediated silencing of DAPK and DUOX1, indicating that RFTS-deleted DNMT1 is a gain of function mutant. Deletion of the 2 different regulatory domains of DNMT1 did not change target preference and chromatin occupancy of DNMT1. In DNMT1-expressing cells, the same target genes were affected with different degree of methylation, which could be attributed to differences in DNA methyltransferase activity.Citation31
Strong alleles of DNMT1 condense chromatin structure at the DAPK and DUOX1 promoters
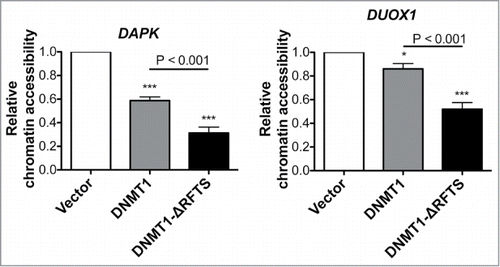
To test whether RFTS and CXXC domains alter the ability of DNMT1 to condense chromatin structure, we analyzed the sensitivity of the DAPK and DUOX1 promoter regions to DNase treatment (). Our data indicate that expression of DNMT1 or DNMT1-ΔRFTS reduced chromatin accessibility at both promoters compared to vector, with DNMT1-ΔRFTS producing the greatest reduction of DNase sensitivity. Thus, silencing of DAPK and DUOX1 is due to DNMT1-dependent alteration of chromatin, and can be increased by deletion of the inhibitory RFTS domain.
Figure 3. DNMT1-ΔRFTS decreases chromatin accessibility at DAPK and DUOX1 promoters. Cells were treated with or without DNA nuclease for 1 hr, prior to detection of promoter DNA by qPCR. The index of chromatin accessibility = 2 ((Ct DNase treated)-(Ct Untreated)). *, p < 0.05; ***, p < 0.001 in comparison to vector cells.
DNA demethylating agent 5-aza-deoxycytidine (5-aza-dC) reverses gene silencing and diminishes the transformation ability of strong DNMT1 alleles
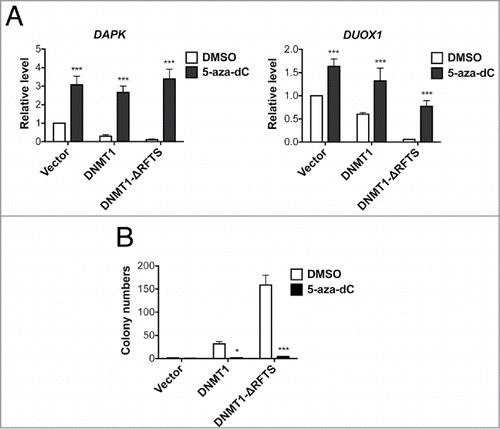
We have demonstrated that the expression of DAPK and DUOX1 is inhibited concurrent with an increase in promoter methylation. We tested whether this methylation-mediated repression is reversible by treating cells with the DNA demethylating agent 5-aza-dC and analyzing expression of DAPK and DUOX1. As expected, 5-aza-dC treatment significantly increased DAPK and DUOX1 levels in DNMT1-expressing cells (). Moreover, 5-aza-dC-treated DNMT1 and DNMT1-ΔRFTS cells completely lost activity in anchorage-independent growth (), indicating that increased methylation-dependent gene regulation is responsible for the oncogenic properties of DNMT1 and DNMT1-ΔRFTS and is reversible.
Figure 4. 5-aza-dC treatment reactivates TSG expression and suppresses DNMT1-dependent transformation. (A) mRNA levels of DAPK (left) and DUOX1 (right) were analyzed by RT-qPCR after 100nM 5-aza-dC treatment for 5 d and normalized to vector cells treated with DMSO. (B) Soft-agar colony formation after 5-aza-dC treatment. ***, p < 0.001 in comparison to the DMSO treated control.
Genome-wide promoter methylation analysis reveals that DNMT1-ΔRFTS cells produce a methylation pattern similar to DNMT1 cells, though more intense
Targeted screening of TSGs indicated that DAPK and DUOX1 promoters are hypermethylated in all DNMT1-expressing cells. DNA from vector, DNMT1 and DNMT1-ΔRFTS cells were used in the high-resolution HpaII tiny fragment enrichment by ligation-mediated PCR (HELP) assay.Citation44 This genomic methylation array of 720,001 probes was designed to focus on CpG islands near transcription start sites. The Pearson Correlation testCitation45 showed that the majority of methylation intensity signals was similar in all cell lines (Fig. S2A). We then analyzed DNA methylation profiles by pairwise comparisons of DNMT1, DNMT1-ΔRFTS and vector control cells. As seen in the volcano plots ( and S2B), there were hyper- and hypomethylated sequences in both DNMT1-expressing cells, with DNMT1-ΔRFTS cells exhibiting greater changes. To systematically analyze methylation targets, we performed Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis to examine the top 1,000 hypermethylated or hypomethylated genes in DNMT1-ΔRFTS cells compared to vector cells (Table S2). Pathway analysis indicated that DNMT1-ΔRFTS drove increased methylation of specific genes involved in cell adhesion, migration and signaling pathways, which are highly related to carcinogenesis. In addition, hypomethylated gene targets in DNMT1-ΔRFTS cells were enriched in lysosomal functions and enzymes in phenylalanine and tyrosine metabolism.
Figure 5. DNMT1-ΔRFTS expression enhances global DNMT1 methylation changes. (A) Genome-wide promoter DNA methylation profiles were obtained using the HELP assay. In volcano plots, the x-axis scores probe-specific methylation ratios and the y-axis scores p-values for the confidence of measurements. The plots allow visualization of methylation differences between vector and DNMT1 cells as well as the differences between vector and DNMT1-ΔRFTS cells. Probes sets that showed significant hyper- or hypomethylation (p < 0.05 for methylation changes (log2(HpaII/MspI)) > 2) are shown in cyan. All other probes are shown in red. (B) Heat map illustration of HpaII-enrichment fragments with methylation changes (log2(HapII/MspI)) > 2 between vector and DNMT1-ΔRFTS cells.
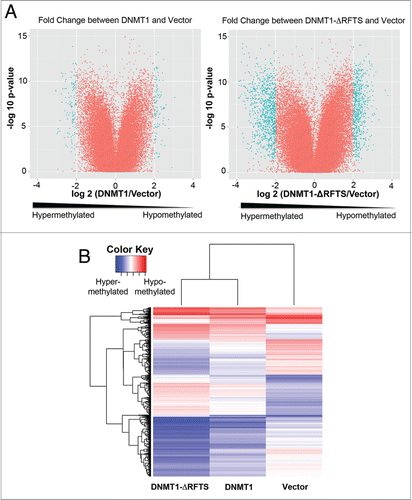
We generated heat maps to compare methylation differences in all 3 sample sets by selecting fragments with more than 4-fold signal changes between DNMT1-ΔRFTS and vector cells (). This big-data visualization analysis showed that DNMT1 and DNMT1-ΔRFTS cells share similar methylation targets and that DNMT1-ΔRFTS cells produces a higher degree of methylation. The DNMT1 and DNMT1-ΔRFTS cells clustered together, suggesting that the RFTS domain does not control locus specificity but limits the degree of DNA modification, just as it limits in vitro activity.Citation31
DNMT1-ΔRFTS cells exhibit genomic hypomethylation
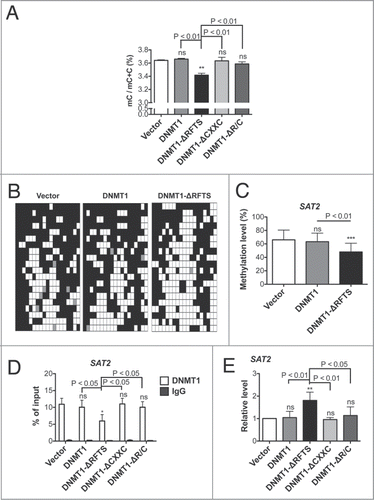
The epigenetic signature of cancer is regional hypermethylation with global hypomethylation. We thus analyzed fractional methyl cytosine content by HPLC ().Citation46 Previous studies have shown that highly overexpressed DNMT1 causes genomic hypermethylation.Citation15-17 However, in our DNMT1 cells in which ectopic expression is at endogenous levels, there was not a significant increase in genomic DNA methylation. In contrast, DNMT1-ΔRFTS cells, which displayed the highest levels of focal hypermethylation, exhibited reduced overall methyl cytosine content in comparison to vector or other DNMT1-expressing cells. We considered that the main site of demethylation might occur in Satellite 2 repeat sequences (SAT2) for 3 reasons. First, the RFTS domain mediates association of DNMT1 to pericentromeric heterochromatin to maintain dense methylationCitation47,48 and SAT2 is the most abundant repeat in the region.Citation49 Second, SAT2-specific hypomethylation has been found in DNMT1-null cellsCitation50,51 and in patients with RFTS-mutated DNMT1.Citation38 Third, DNA hypomethylation and RNA up-regulation of SAT2 are highly associated with various cancers and contributes to genomic instability.Citation49 To test whether SAT2 is hypomethylated in our cell lines, we performed bisulfite sequencing (). SAT2 methylation was significant reduced from 66% (vector) and 63% (DNMT1) to 48% in DNMT1-ΔRFTS cells. Further, we analyzed chromatin occupancy of DNMT1 on SAT2 loci by DNMT1 ChIP (). The data indicate that DNMT1-ΔRFTS expression reduced association between DNMT1 and SAT2 loci. Unlike what was observed for SAT2, there was no significant methylation change in LINE1 DNA repeats (Fig. S3). Therefore, specific demethylation of SAT2 might be due to the impaired association between DNMT1-ΔRFTS and the pericentromeric region. Our results suggest that demethylation of SAT2 and promoter hypomethylation detected in the HELP assay might both contribute to reduced genomic methylation observed in DNMT1-ΔRFTS cells. Moreover, we investigated whether SAT2 DNA hypomethylation is associated with transcription. Indeed, expression of SAT2 non-coding RNA was increased in DNMT1-ΔRFTS cells, but was not altered in cell lines with alleles of DNMT1 that are weaker ().
Figure 6. Genomic hypomethylation is found in DNMT1-ΔRFTS cells. (A) 5-methylcytosine (mC) content of the total cytosine pool was determined by HPLC. (B) Bisulfite sequencing of SAT2. White squares represent unmethylated CpGs, black squares represent methylated CpGs, and gray squares represent undetermined sites. Each row is an independent sequencing result. (C) Quantitation of SAT2 bisulfite sequencing. (D) DNMT1 chromatin occupancy was analyzed using DNMT1 ChIP and qPCR. (E) Expression of SAT2 non-coding RNA was analyzed by RT-qPCR and normalized to vector cells. *, p < 0.05; **, p < 0.01; ***, p < 0.001 in comparison to vector cells.
DNMT1-ΔRFTS expression has similar effects in H358 lung cancer cells
To rule out a cell-specific effect, we established stable cell lines in H358 cells to determine the effect of DNMT1-ΔRFTS expression in malignant cells (Fig. S4A). DNMT1-ΔRFTS expression enhanced the proliferation, invasion and soft-agar colony growth of the cells, while full-length DNMT1 expression behaved similarly to vector control cells (Fig. S4B-D). Moreover, ectopic expression of DNMT1-ΔRFTS slightly inhibited expression of DAPK and DUOX1 (Fig. S5A). Although DNMT1-ΔRFTS cells did not express significantly more SAT2 RNA transcripts (Fig. S5A), there was a notable methylation reduction of SAT2 in DNMT1-ΔRFTS cells (Fig. S5B and C). These data suggest that the biological function of DNMT1-ΔRFTS is not cell-specific.
Discussion
We proposed that the RFTS domain is inhibitory by virtue of DNA-competitive association with the DNMT1 active site,Citation31 while other data suggested that the CXXC domain is DNA-competitive and inhibitory.Citation37 Here, we hypothesized that expression of hyperactive DNMT1 lacking an autoinhibitory domain could enhance transformation by altering DNA methylation. Thus, we modestly expressed full-length and deletion forms of DNMT1 in immortalized HBEC3 cells to examine their oncogenic potential and alteration in DNA methylation. Full-length DNMT1 expression triggered cellular transformation while DNMT1-ΔRFTS functioned as a stronger oncoprotein. The oncogenic effects of DNMT1-ΔRFTS depended on the presence of the CXXC domain, which is apparently a positive factor. Deletion of either regulatory domain resulted in the same apparent target preference; expression of all DNMT1 alleles increased methylation of DAPK and DUOX1 promoters. DNMT1 and DNMT1-ΔRFTS cells share similar hypermethylated targets in genome-wide analysis as well, though deletion of RFTS increased the degree of DNA methylation.
Given previous findings that overexpressed or activated DNMT1 caused non-specific genomic hypermethylation,Citation15-17 we surprisingly discovered that DNMT1-ΔRFTS expression at endogenous levels led to demethylation of SAT2 and in the genome. This finding is consistent with a previous study on RFTS-mutated DNMT1,Citation38 in which point mutations in the RFTS domain caused SAT2 and genomic hypomethylation. The study also showed that mutations in the RFTS domain of DNMT1 impaired binding with heterochromatin.Citation38 We confirmed the impaired association between DNMT1 and SAT2 loci in DNMT1-ΔRFTS cells by DNMT1 ChIP. Because DNMT1 may function as an oligomeric complex,Citation52 RFTS-deleted DNMT1 and endogenous DNMT1 may form a heterooligomer that is impaired in association with pericentromeric heterochromatin. Thus, we suggest a model in which deletion of the RFTS domain activates DNMT1 for euchromatic DNA-binding, but decreases chromatin occupancy of DNMT1 to heterochromatic SAT2 loci by virtue of a missing protein interaction, leading to passive DNA demethylation (). Searching the catalog of somatic mutation in cancer (COSMIC) database, 26 mutation sites within the RFTS domain of DNMT1 were found (Table S3). These DNMT1 mutants might promote malignancy by increasing DNA methyltransferase binding and activity on euchromatic DNA while being disadvantaged in pericentromeric SAT2 association and methylation.
Figure 7. Dual roles for RFTS domain in DNMT1-dependent DNA methylation. (A) RFTS-targeted DNMT1 associated proteins (RAP) are proposed to relieve inhibition of DNMT1 for access to euchromatin. (B) The RFTS domain mediates association between DNMT1 and pericentromeric heterochromatin. (C) In cancer, overexpression of RAPs or mutation of RFTS is proposed to relieve DNMT1 inhibition, thereby increasing methylation and silencing of TSGs. However, because the RFTS domain is required for association with heterochromatic SAT2 sequences, DNMT1 with mutant RFTS may be less associated with such sequences, accounting for global hypomethylation.
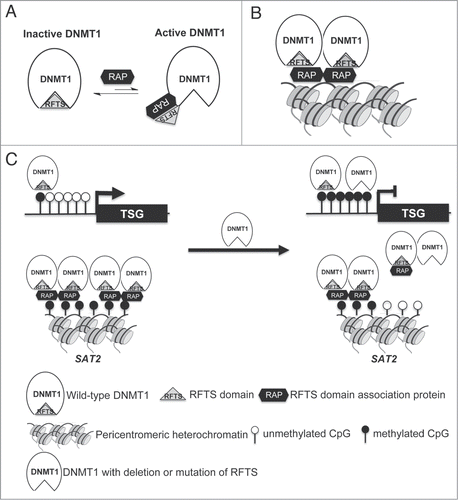
RFTS-targeted DNMT1 associated proteins (RAPs) are likely to participate in these mechanisms. To our knowledge, there are at least 3 known RAPs including ubiquitin-like containing PHD and RING finger domain protein 1 (UHRF1),Citation29,30,35 ubiquitin-specific-processing protease 7 (USP7)Citation53 and N-α-acetyltransferase 10 NatA catalytic subunit (NAA10).Citation54 These proteins have been shown to recruit DNMT1 to specific loci and stimulate its methylation activity, causing site-specific hypermethylation. Moreover, these proteins were found up-regulated in lung cancers,Citation54–56 suggesting that release of RFTS domain inhibition might drive cancer formation via hypermethylayion. If this is the case, targeting of these binding partners could be a promising therapeutic strategy to limit DNMT1-dependent hypermethylation in cancer.
Because of the link between hyperactive DNMT1 and transcriptional repression of TSGs, DNMT1-mediated DNA hypermethylation is emerging as a crucial therapeutic target.Citation57,58 The current approach is to inhibit expression or hyperactivity of DNMT1.Citation59-61 However, demethylating agents lead to unavoidable non-specific genomic demethylation causing genomic instability, DNA damage, oncogene reactivation, and promote opportunities for cancer progression.Citation62-65 Because the RFTS domain functions as a key regulator of DNMT1 function, targeting RFTS interactions may revert euchromatin-associated DNMT1 activation while also normalizing pericentromeric DNA methylation.
In conclusion, our study reveals the functional roles of the RFTS domain of DNMT1 in maintenance of a nontransformed epigenome. We have demonstrated that deletion of RFTS enhanced the oncogenic potential of DNMT1 by increasing promoter methylation of TSGs such as DAPK and DUOX1. However, DNMT1-ΔRFTS also decreased association of DNMT1 with the pericentromeric region, causing SAT2 demethylation. Because DNMT1-ΔRFTS was able to reprogram the overall methylation pattern of epithelial cells in a manner that is common in cancer, the data suggest that RFTS may be a target of tumor-specific dysregulation.
Materials and Methods
Cell Culture
HBEC3 and stable cell lines were grown in KSFM media supplemented with bovine pituitary extract and recombinant human EGF. H358 cells (CRL-5807, ATCC) and stable cell lines were grown in RPMI1640 media with 10% serum.
Establishment of Stable Cell Lines
To establish full length or deletion mutant human DNMT1 stable lines, a full-length DNMT1 cDNA clone (SC325419, Origene) was used as template to amplify full-length or deletion mutant DNMT1 fragments. PCR fragments were first T/A cloned into pGEM-T easy vector (A1360, Promega) and then subcloned into pLenti6/V5 vector (K4950–00, Invitrogen). Viral production and transduction was performed using the ViralPower Bsd Lentiviral Support Kit (K4950–00, Invitrogen). Primer pairs used for plasmid constructions are provided in Table S4.
Proliferation and Invasion Assay
To analyze proliferation, 1,000 (HBEC3) or 5,000 (H358) cells were seeded in replicates of 6 in KSFM media with or without EGF supplementation in 96-well plates. Relative cell numbers were analyzed using Resazurin (R7017, Sigma) 72 hrs after seeding. Data were collected from 4 independent experiments. Cell invasion was analyzed using the CultreCoat® 24 Well Low BME Invasion Assay kit (3481–024–01, Trevigen).
RT-qPCR
Total RNA was extracted using the RNeasy Mini Kit (74106, Qiagen) with DNase treatment (79254, Qiagen) to eliminate DNA contamination. Equal amounts of RNA were reverse transcribed to generate cDNA using the QuantiTect Reverse Transcription Kit (205314, Qiagen). Specific primer pairs were then used to amplify targets (Table S4). qPCR reactions were conducted with iQ SYBR Green Supermix (170–8884, Bio-Rad). All data were collected from 3 or 4 independent experiments.
Immunoblotting
Protein extracts from each stable cell line were prepared in RIPA buffer (89900, Thermo Scientific) according to the manufacturer's instructions. Equal amounts of protein were separated using NuPAGE® Novex® 3–8% Tris-Acetate Gels and transferred to 0.2 μm nitrocellulose membranes at 4°C overnight. DNMT1 (WH0001786M1, Sigma) and actin (ab3280, Abcam) were detected using specific antibodies and visualized by SuperSignal West Femto Substrate (34096, Thermo Scientific).
Adherent and soft-agar colony formation
For adherent colony formation, 200 cells were seeded on 10-cm culture dishes in KSFM media without EGF, and allowed to grow for 12 days, followed by 4% methylene blue (M9140, Sigma) staining. Colonies >3 mm were counted. For soft-agar colony formation, 10,000 (HBEC3) or 5,000 (H358) cells were resuspended in media with 0.4% agarose and plated over a layer of 0.6 % agarose. Cells were incubated at 37°C for 6 (HBEC3) or 4 (H358) weeks and then colonies were stained with MTT (M5655, Sigma). Colony images were acquired with ChemiDoc XRS (Bio-Rad) and quantified using Quantity One software (Bio-Rad). All data were collected from 2 or 3 independent experiments, each performed in triplicate.
Methylation assay
Genomic DNA was extracted using DNeasy Blood & Tissue Kit (69506, Qiagen). Promoter methylation analysis was performed using MethylMiner Methylated DNA Enrichment Kit (ME10025, Invitrogen) or bisulfite sequencing. For MethylMiner experiments, genomic DNA was first fragmented by sonication to an average size of 400 bp. Methylated DNA was captured following the manufacturer protocol. The methylation level was analyzed using specific primer sets with qPCR (Table S4). Ten percent of input DNA was used as a control. Data were collected from 3 independent experiments. For bisulfite sequencing, genomic DNA was treated with bisulfite using EpiTect Bisulfite kit (59124, Qiagen). Bisulfite treated DNA was then used as a template and PCR was performed using specific primer pairs (Table S4). Final PCR products were gel purified and cloned into the pGEM-T easy vector (A1360, Promega). Independent clones were subjected to sequencing. Global methylation was analyzed by HPLC. Genomic DNA was digested to nucleosides using DNA Degradase Plus Kit (E2021, Zymo Research). One μg digested DNA was separated on a 25 cm × 4.6 μm C18 Supelco column using 7.5 mM ammonium phosphate and methanol. Retention time of each nucleoside was determined by nucleotide standards. Data were collected from 3 independent experiments, each performed in duplicate.
ChIP
ChIP was performed with Magna ChIP HiSens chromatin immunoprecipitation kit (17–10461, Millipore), DNMT1 antibody (IMG-261A, Imgenex), and analyzed using qPCR. Ten percent of input DNA was used as a control. All data were collected from 3 independent experiments.
Nuclease-protection assay
Chromatin accessibility was assessed using EpiQ™ Chromatin Preparation Kit (172–5402, Bio-Rad). Primers used are listed in Table S4. All data were collected from 3 independent experiments, each performed in triplicate.
HELP assay and data analysis
Genomic DNA was extracted using the DNeasy Blood & Tissue Kit (69506, Qiagen). The HELP assay was conducted in the Epigenomics Core Facility of Weill Cornell Medical College. Samples were hybridized to a custom oligonucleotide array (Roche NimbleGen, Madision, WI; design name: 100128_HG19_MKF_HELP_ChIP_HX3). HELP data were processed using the HELP package in R from Bioconductor. Primary data are available from the NCBI GEO public database (accession number: GSE57829).
Statistical analysis
All data were presented as mean ± SD. One-way ANOVA was used to calculate P-values and determine significance. P-values lower than 0.05 were considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
950886_SupplementaryFiguresTables.docx
Download MS Word (1.3 MB)Acknowledgments
The HBEC3 line was a kind gift of Dr. John Minna. This work was supported by grant R01CA075954 and contract HHSN261200433000C from the National Cancer Institute awarded to CB.
Supplemental Materials
Supplemental data for this article can be accessed on the publisher's website.
References
- Robertson KD. DNA methylation and human disease. Nat Rev Genet 2005; 6:597-610; PMID:16136652; http://dx.doi.org/10.1038/nrg1655
- Issa J-P. CpG island methylator phenotype in cancer. Nat Rev Cancer 2004; 4:988-93; PMID:15573120
- Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer 2004; 4:707-17; PMID:15343277; http://dx.doi.org/10.1038/nrc1432
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415-28; PMID:12042769
- Feinberg AP, Gehrke CW, Kuo KC, Ehrlich M. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res 1988; 48:1159-61; PMID:3342396
- Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003; 300:455; PMID:12702868; http://dx.doi.org/10.1126/science.1083557
- Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene 2002; 21:5400-13; PMID:12154403; http://dx.doi.org/10.1038/sj.onc.1205651
- Christman JK, Sheikhnejad G, Marasco CJ, Sufrin JR. 5-Methyl-2'-deoxycytidine in single-stranded DNA can act in cis to signal de novo DNA methylation. Proc Natl Acad Sci USA 1995; 92:7347-51; PMID:7638194; http://dx.doi.org/10.1073/pnas.92.16.7347
- Pradhan S, Talbot D, Sha M, Benner J, Hornstra L, Li E, Jaenisch R, Roberts RJ. Baculovirus-mediated expression and characterization of the full-length murine DNA methyltransferase. Nucleic Acids Res 1997; 25:4666-73; PMID:9358180; http://dx.doi.org/10.1093/nar/25.22.4666
- Jair K-W, Bachman KE, Suzuki H, Ting AH, Rhee I, Yen R-WC, Baylin SB, Schuebel KE. De novo CpG island methylation in human cancer cells. Cancer Res 2006; 66:682-92; PMID:16423997; http://dx.doi.org/10.1158/0008-5472.CAN-05-1980
- el-Deiry WS, Nelkin BD, Celano P, Yen RW, Falco JP, Hamilton SR, Baylin SB. High expression of the DNA methyltransferase gene characterizes human neoplastic cells and progression stages of colon cancer. Proc Natl Acad Sci USA 1991; 88:3470-4; PMID:2014266; http://dx.doi.org/10.1073/pnas.88.8.3470
- Belinsky SA, Nikula KJ, Baylin SB, Issa JP. Increased cytosine DNA-methyltransferase activity is target-cell-specific and an early event in lung cancer. Proc Natl Acad Sci USA 1996; 93:4045-50; PMID:8633014; http://dx.doi.org/10.1073/pnas.93.9.4045
- Lin R-K, Hsu H-S, Chang J-W, Chen C-Y, Chen J-T, Wang Y-C. Alteration of DNA methyltransferases contributes to 5'CpG methylation and poor prognosis in lung cancer. Lung Cancer 2007; 55:205-13; PMID:17140695; http://dx.doi.org/10.1016/j.lungcan.2006.10.022
- Kimura F, Seifert H-H, Florl AR, Santourlidis S, Steinhoff C, Swiatkowski S, Mahotka C, Gerharz C-D, Schulz WA. Decrease of DNA methyltransferase 1 expression relative to cell proliferation in transitional cell carcinoma. Int J Cancer 2003; 104:568-78; PMID:12594811; http://dx.doi.org/10.1002/ijc.10988
- Wu J, Issa JP, Herman J, Bassett DE, Nelkin BD, Baylin SB. Expression of an exogenous eukaryotic DNA methyltransferase gene induces transformation of NIH 3T3 cells. Proc Natl Acad Sci USA 1993; 90:8891-5; PMID:8415627; http://dx.doi.org/10.1073/pnas.90.19.8891
- Bakin AV, Curran T. Role of DNA 5-methylcytosine transferase in cell transformation by fos. Science 1999; 283:387-90; PMID:9888853; http://dx.doi.org/10.1126/science.283.5400.387
- Biniszkiewicz D, Gribnau J, Ramsahoye B, Gaudet F, Eggan K, Humpherys D, Mastrangelo M-A, Jun Z, Walter J, Jaenisch R. Dnmt1 overexpression causes genomic hypermethylation, loss of imprinting, and embryonic lethality. Mol Cell Biol 2002; 22:2124-35; PMID:11884600; http://dx.doi.org/10.1128/MCB.22.7.2124-2135.2002
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992; 69:915-26; PMID:1606615; http://dx.doi.org/10.1016/0092-8674(92)90611-F
- Gaudet F. Induction of tumors in mice by genomic hypomethylation. Science 2003; 300:489-92; PMID:12702876; http://dx.doi.org/10.1126/science.1083558
- Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, Fuks F, Coco Lo F, Kouzarides T, Nervi C, et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 2002; 295:1079-82; PMID:11834837; http://dx.doi.org/10.1126/science.1065173
- Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden J-M, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006; 439:871-4; http://dx.doi.org/10.1038/nature04431
- Esteve PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR, Carey MF, Pradhan S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev 2006; 20:3089-103; PMID:17085482; http://dx.doi.org/10.1101/gad.1463706
- Smallwood A, Esteve PO, Pradhan S, Carey M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev 2007; 21:1169-78; PMID:17470536; http://dx.doi.org/10.1101/gad.1536807
- Chuang LS. Human DNA-(Cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science 1997; 277:1996-2000; PMID:9302295; http://dx.doi.org/10.1126/science.277.5334.1996
- Hervouet E, Lalier L, Debien E, Cheray M, Geairon A, Rogniaux H, Loussouarn D, Martin SA, Vallette FM, Cartron P-F. Disruption of Dnmt1/PCNA/UHRF1 interactions promotes tumorigenesis from human and mice glial cells. PLoS ONE 2010; 5:e11333; PMID:20613874; http://dx.doi.org/10.1371/journal.pone.0011333
- Pradhan S, Kim G-D. The retinoblastoma gene product interacts with maintenance human DNA (cytosine-5) methyltransferase and modulates its activity. EMBO J 2002; 21:779-88; PMID:11847125; http://dx.doi.org/10.1093/emboj/21.4.779
- Leonhardt H, Page AW, Weier HU, Bestor TH. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 1992; 71:865-73; PMID:1423634; http://dx.doi.org/10.1016/0092-8674(92)90561-P
- Margot JB, Aguirre-Arteta AM, Di Giacco BV, Pradhan S, Roberts RJ, Cardoso MC, Leonhardt H. Structure and function of the mouse DNA methyltransferase gene: Dnmt1 shows a tripartite structure. J Mol Biol 2000; 297:293-300; PMID:10715201; http://dx.doi.org/10.1006/jmbi.2000.3588
- Bostick M, Kim JK, Estève P-O, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007; 317:1760-4; PMID:17673620; http://dx.doi.org/10.1126/science.1147939
- Sharif J, Muto M, Takebayashi S-I, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T, Okamura K, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007; 450:908-12; PMID:17994007; http://dx.doi.org/10.1038/nature06397
- Syeda F, Fagan RL, Wean M, Avvakumov GV, Walker JR, Xue S, Dhe-Paganon S, Brenner C. The replication focus targeting sequence (RFTS) domain is a DNA-competitive inhibitor of Dnmt1. J Biol Chem 2011; 286:15344-51; PMID:21389349; http://dx.doi.org/10.1074/jbc.M110.209882
- Takeshita K, Suetake I, Yamashita E, Suga M, Narita H, Nakagawa A, Tajima S. Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1). Proc Natl Acad Sci USA 2011; 108:9055-9; PMID:21518897; http://dx.doi.org/10.1073/pnas.1019629108
- Hashimoto H, Liu Y, Upadhyay AK, Chang Y, Howerton SB, Vertino PM, Zhang X, Cheng X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res 2012; 40:4841-9; PMID:22362737; http://dx.doi.org/10.1093/nar/gks155
- Berkyurek AC, Suetake I, Arita K, Takeshita K, Nakagawa A, Shirakawa M, Tajima S. The DNA methyltransferase Dnmt1 directly interacts with the SET and RING finger-associated (SRA) domain of the multifunctional protein Uhrf1 to facilitate accession of the catalytic center to hemi-methylated DNA. J Biol Chem 2014; 289:379-86; PMID:24253042; http://dx.doi.org/10.1074/jbc.M113.523209
- Bashtrykov P, Jankevicius G, Jurkowska RZ, Ragozin S, Jeltsch A. The UHRF1 protein stimulates the activity and specificity of the maintenance DNA methyltransferase DNMT1 by an allosteric mechanism. J Biol Chem 2014; 289:4106-15; PMID:24368767; http://dx.doi.org/10.1074/jbc.M113.528893
- Bashtrykov P, Rajavelu A, Hackner B, Ragozin S, Carell T, Jeltsch A. Targeted mutagenesis results in an activation of DNA methyltransferase 1 and confirms an autoinhibitory role of its RFTS domain. Chembiochem 2014; 15:743-8; PMID:24532244; http://dx.doi.org/10.1002/cbic.201300740
- Song J, Rechkoblit O, Bestor TH, Patel DJ. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 2011; 331:1036-40; PMID:21163962; http://dx.doi.org/10.1126/science.1195380
- Klein CJ, Botuyan M-V, Wu Y, Ward CJ, Nicholson GA, Hammans S, Hojo K, Yamanishi H, Karpf AR, Wallace DC, et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet 2011; 43:595-600; PMID:21532572; http://dx.doi.org/10.1038/ng.830
- Sato M, Vaughan MB, Girard L, Peyton M, Lee W, Shames DS, Ramirez RD, Sunaga N, Gazdar AF, Shay JW, et al. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res 2006; 66:2116-28; PMID:16489012; http://dx.doi.org/10.1158/0008-5472.CAN-05-2521
- Damiani LA, Yingling CM, Leng S, Romo PE, Nakamura J, Belinsky SA. Carcinogen-induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cells. Cancer Res 2008; 68:9005-14; PMID:18974146; http://dx.doi.org/10.1158/0008-5472.CAN-08-1276
- Kim DH, Nelson HH, Wiencke JK, Christiani DC, Wain JC, Mark EJ, Kelsey KT. Promoter methylation of DAP-kinase: association with advanced stage in non-small cell lung cancer. Oncogene 2001; 20:1765-70; PMID:11313923; http://dx.doi.org/10.1038/sj.onc.1204302
- Pulling LC, Vuillemenot BR, Hutt JA, Devereux TR, Belinsky SA. Aberrant promoter hypermethylation of the death-associated protein kinase gene is early and frequent in murine lung tumors induced by cigarette smoke and tobacco carcinogens. Cancer Res 2004; 64:3844-8; PMID:15172992; http://dx.doi.org/10.1158/0008-5472.CAN-03-2119
- Luxen S, Belinsky SA, Knaus UG. Silencing of DUOX NADPH oxidases by promoter hypermethylation in lung cancer. Cancer Res 2008; 68:1037-45; PMID:18281478; http://dx.doi.org/10.1158/0008-5472.CAN-07-5782
- Khulan B. Comparative isoschizomer profiling of cytosine methylation: The HELP assay. Genome Res 2006; 16:1046-55; PMID:16809668; http://dx.doi.org/10.1101/gr.5273806
- Thompson RF, Reimers M, Khulan B, Gissot M, Richmond TA, Chen Q, Zheng X, Kim K, Greally JM. An analytical pipeline for genomic representations used for cytosine methylation studies. Bioinformatics 2008; 24:1161-7; PMID:18353789; http://dx.doi.org/10.1093/bioinformatics/btn096
- Magaña AA, Wrobel K, Caudillo YA, Zaina S, Lund G, Wrobel K. High-performance liquid chromatography determination of 5-methyl-2“-deoxycytidine, 2-”deoxycytidine, and other deoxynucleosides and nucleosides in DNA digests. Anal Biochem 2008; 374:378-85; http://dx.doi.org/10.1016/j.ab.2007.11.026
- Easwaran HP, Schermelleh L, Leonhardt H, Cardoso MC. Replication-independent chromatin loading of Dnmt1 during G2 and M phases. EMBO Rep 2004; 5:1181-6; PMID:15550930; http://dx.doi.org/10.1038/sj.embor.7400295
- Schneider K, Fuchs C, Dobay A, Rottach A, Qin W, Wolf P, Alvarez-Castro JM, Nalaskowski MM, Kremmer E, Schmid V, et al. Dissection of cell cycle-dependent dynamics of Dnmt1 by FRAP and diffusion-coupled modeling. Nucleic Acids Res 2013; 41:4860-76; PMID:23535145; http://dx.doi.org/10.1093/nar/gkt191
- Ting DT, Lipson D, Paul S, Brannigan BW, Akhavanfard S, Coffman EJ, Contino G, Deshpande V, Iafrate AJ, Letovsky S, et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 2011; 331:593-6; PMID:21233348; http://dx.doi.org/10.1126/science.1200801
- Rhee I, Jair KW, Yen RW, Lengauer C, Herman JG, Kinzler KW, Vogelstein B, Baylin SB, Schuebel KE. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature 2000; 404:1003-7; PMID:10801130; http://dx.doi.org/10.1038/35010000
- Espada J. Human DNA methyltransferase 1 is required for maintenance of the histone H3 modification pattern. J Biol Chem 2004; 279:37175-84; PMID:15220328; http://dx.doi.org/10.1074/jbc.M404842200
- Fellinger K, Rothbauer U, Felle M, Längst G, Leonhardt H. Dimerization of DNA methyltransferase 1 is mediated by its regulatory domain. J Cell Biochem 2009; 106:521-8; PMID:19173286; http://dx.doi.org/10.1002/jcb.22071
- Felle M, Joppien S, Nemeth A, Diermeier S, Thalhammer V, Dobner T, Kremmer E, Kappler R, Langst G. The USP7/Dnmt1 complex stimulates the DNA methylation activity of Dnmt1 and regulates the stability of UHRF1. Nucleic Acids Res 2011; 39:8355-65; PMID:21745816; http://dx.doi.org/10.1093/nar/gkr528
- Lee C-F, Ou DS-C, Lee S-B, Chang L-H, Lin R-K, Li Y-S, Upadhyay AK, Cheng X, Wang Y-C, Hsu H-S, et al. hNaa10p contributes to tumorigenesis by facilitating DNMT1-mediated tumor suppressor gene silencing. J Clin Invest 2010; 120:2920-30; PMID:20592467; http://dx.doi.org/10.1172/JCI42275
- Unoki M, Daigo Y, Koinuma J, Tsuchiya E, Hamamoto R, Nakamura Y. UHRF1 is a novel diagnostic marker of lung cancer. Br J Cancer 2010; 103:217-22; PMID:20517312; http://dx.doi.org/10.1038/sj.bjc.6605717
- Daskalos A, Oleksiewicz U, Filia A, Nikolaidis G, Xinarianos G, Gosney JR, Malliri A, Field JK, Liloglou T. UHRF1-mediated tumor suppressor gene inactivation in nonsmall cell lung cancer. Cancer 2011; 117:1027-37; PMID:21351083; http://dx.doi.org/10.1002/cncr.25531
- Laird PW, Jackson-Grusby L, Fazeli A, Dickinson SL, Jung WE, Li E, Weinberg RA, Jaenisch R. Suppression of intestinal neoplasia by DNA hypomethylation. Cell 1995; 81:197-205; PMID:7537636; http://dx.doi.org/10.1016/0092-8674(95)90329-1
- Suzuki M, Sunaga N, Shames DS, Toyooka S, Gazdar AF, Minna JD. RNA interference-mediated knockdown of DNA methyltransferase 1 leads to promoter demethylation and gene re-expression in human lung and breast cancer cells. Cancer Res 2004; 64:3137-43; PMID:15126351; http://dx.doi.org/10.1158/0008-5472.CAN-03-3046
- Ramchandani S, MacLeod AR, Pinard M, Hofe von E, Szyf M. Inhibition of tumorigenesis by a cytosine-DNA, methyltransferase, antisense oligodeoxynucleotide. Proc Natl Acad Sci USA 1997; 94:684-9; PMID:9012845; http://dx.doi.org/10.1073/pnas.94.2.684
- McCabe MT, Low JA, Daignault S, Imperiale MJ, Wojno KJ, Day ML. Inhibition of DNA methyltransferase activity prevents tumorigenesis in a mouse model of prostate cancer. Cancer Res 2006; 66:385-92; PMID:16397253; http://dx.doi.org/10.1158/0008-5472.CAN-05-2020
- Datta J, Ghoshal K, Denny WA, Gamage SA, Brooke DG, Phiasivongsa P, Redkar S, Jacob ST. A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res 2009; 69:4277-85; PMID:19417133; http://dx.doi.org/10.1158/0008-5472.CAN-08-3669
- Szyf M. Targeting DNA methylation in cancer. Ageing Res Rev 2003; 2:299-328; PMID:12726777; http://dx.doi.org/10.1016/S1568-1637(03)00012-6
- Loriot A. Transient down-regulation of DNMT1 methyltransferase leads to activation and stable hypomethylation of MAGE-A1 in melanoma cells. J Biol Chem 2006; 281:10118-26; PMID:16497664; http://dx.doi.org/10.1074/jbc.M510469200
- Yaqinuddin A, Qureshi SA, Qazi R, Farooq S, Abbas F. DNMT1 silencing affects locus specific DNA methylation and increases prostate cancer derived PC3 cell invasiveness. J Urol 2009; 182:756-61; PMID:19539327; http://dx.doi.org/10.1016/j.juro.2009.03.082
- Morey Kinney SR, Moser MT, Pascual M, Greally JM, Foster BA, Karpf AR. Opposing roles of Dnmt1 in early- and late-stage murine prostate cancer. Mol Cell Biol 2010; 30:4159-74; PMID:20584988; http://dx.doi.org/10.1128/MCB.00235-10