
Abstract
Regeneration of skeletal muscle relies on a population of quiescent stem cells (satellite cells) and is impaired in very old (geriatric) individuals undergoing sarcopenia. Stem cell function is essential for organismal homeostasis, providing a renewable source of cells to repair damaged tissues. In adult organisms, age-dependent loss-of-function of tissue-specific stem cells is causally related with a decline in regenerative potential. Although environmental manipulations have shown good promise in the reversal of these conditions, recently we demonstrated that muscle stem cell aging is, in fact, a progressive process that results in persistent and irreversible changes in stem cell intrinsic properties. Global gene expression analyses uncovered an induction of p16INK4a in satellite cells of physiologically aged geriatric and progeric mice that inhibits satellite cell-dependent muscle regeneration. Aged satellite cells lose the repression of the INK4a locus, which switches stem cell reversible quiescence into a pre-senescent state; upon regenerative or proliferative pressure, these cells undergo accelerated senescence (geroconversion), through Rb-mediated repression of E2F target genes. p16INK4a silencing rejuvenated satellite cells, restoring regeneration in geriatric and progeric muscles. Thus, p16INK4a/Rb-driven stem cell senescence is causally implicated in the intrinsic defective regeneration of sarcopenic muscle. Here we discuss on how cellular senescence may be a common mechanism of stem cell aging at the organism level and show that induction of p16INK4a in young muscle stem cells through deletion of the Polycomb complex protein Bmi1 recapitulates the geriatric phenotype.
Stem Cell Aging
Adult or somatic stem cells represent a small population of long-lived progenitor cells which reside in an undifferentiated state within most or all fully differentiated adult tissues and whose function is fundamental for maintaining tissue homeostasis and for mediating growth and regeneration after damage or in disease. It has been postulated that aging of an organism is in great part a direct result of stem cell loss-of-function. With age there is both a progressive decline in the stem cell self-renewal ability, causing in some cases a depletion of the stem cell pool, and a loss of activation/proliferative potential, resulting in impairment of the regenerative process due to an inability to generate the appropriate progeny.Citation1,2 Tissues vary in their homeostatic and regenerative requirements and that is reflected on the turnover of their associated stem cell populations.Citation3,4 The heterogeneity in stem cell differentiation and proliferative potential among different tissues underlay different susceptibilities to age-associated stressors throughout the organism lifespan. Stem cells of tissues with high cellular turnover, such as the intestine or the haematopoietic system, will be subjected to genomic instability that is associated with high rates of DNA replication. By contrast, low cellular turnover tissues, such as the skeletal muscle and the cerebral cortex, contain stem cell populations that are for the most part maintained in a quiescent state and only induced to proliferate upon injury or stress. These non-dividing cells undergo a progressive decline in function with chronological aging, although the mechanism behind this process is poorly understood.Citation5-7 Cellular senescence corresponds to a state of irreversible cellular arrest and can be triggered in primary cells by different stimuli, including expression of activated oncogenes (oncogene-induced senescence), or serial passaging (replicative senescence). Specifically, accelerated cellular senescence (i.e. geroconversion) is believed to occur when cellular growth is promoted while cell cycle progression is inhibited.Citation8,9 The concept of cellular senescence as a driving force for age related pathologies was first introduced by Hayflick and Moorhead in 1961 and the hypothesis has sustained until the present without a definitive prove.Citation10 Meanwhile, senescent cells have indeed been observed during organismal agingCitation11-18 as well as in other different physiological contexts, including embryonic development,Citation19,20 tissue repairCitation21,22 and tumor suppression.Citation23 The physiological role of senescence in these contexts has also been addressed and it involves functions that range from tissue remodeling during development to limiting excessive tissue fibrosis during wound healing and tissue repair.Citation24,25 However, how do senescent cells promote age-related tissue dysfunction is a question that remains to be answered. One hypothesis is that senescence acts on stem cells to limit tissues regenerative potential during aging. Some hints of such mechanism have been observed in skeletal muscle and fat tissue from BubR1 progeroid mice,Citation26 where senescence markers co-localized with resident stem cells.
The resident adult stem cells of skeletal muscle are termed satellite cells since they are located between the myofibers and the basal lamina. Satellite cells express the paired box transcription factor Pax7 and normally reside in a quiescent state until activated by environmental signals (e.g., injury or stress) or in pathological environments (e.g., in degenerative muscle diseases).Citation27-29 Following activation, satellite cells originate 2 types of progeny: self-renewed satellite cells which maintain the stem cell pool, and committed myogenic cells, which form new muscle fibers where necessary to sustain postnatal muscle repair and growth.Citation30 Satellite cell aging is characterized by a loss of regenerative potential,Citation31,32 which is particularly pronounced in sarcopenic muscles.Citation33 This is mostly a consequence of impairments in activation and proliferative potential in response to injury, which account for the primary age-specific defects in muscle repair. The regenerative defects are also associated with a delayed inflammation and an increase in tissue fibrosis and aged satellite cells display a tendency to enter alternative differentiation programs by adopting fibroblastic and adipogenic fates.Citation34-36
Given the negative impact of satellite cell aging on muscle regeneration, there has been an extensive research effort in characterizing the origin of the aging phenotypes, focusing on strategies of rejuvenation. The prevalent view has been that satellite-cell-dependent muscle regeneration decline with aging is mostly due to extrinsic factors and can therefore be reversed by exposure to an external youthful environment.Citation2 This view originates in heterochronic parabiosis and hetero-grafting studies between young and old (non-geriatric) mice. Several labs have shown that the regenerative capacity of injured muscle tissue from old mice can be enhanced to match the efficiency of young mice when their blood circulation is joined.Citation34,37,38 This is mostly a consequence of satellite cell rejuvenation by exposure to young serum, leading to the re-establishment of proliferative and myogenic potential necessary to regenerate the injured muscle.Citation38 These data supported earlier hetero-grafting studiesCitation39-43 and suggest that there are factors that accumulate either systemically or in the niche of old animals, which are deleterious to the satellite cell and regenerative process. More importantly, it suggests that the old serum/niche is depleted of factors that are present in the blood of young animals, which could be therefore sufficient to enhance regenerative potential of the injured skeletal muscle and can rejuvenate old (non-geriatric) satellite cells. Several niche-derived and systemic factors responsible for the regenerative decline in old animals have been identified, including Transforming growth factor β (TGFβ) and Fibroblast Growth Factor 2 (FGF2) from the niche,Citation32,44,45 as well as systemically derived Wnt signaling.Citation34 These studies focused on pathways which are over-activated in aging and proposed rejuvenation strategies based on inhibition of these signaling cascades. Two recent studies, however, have identified systemic derived factors capable of reversing age-related impairment of muscle regeneration and rejuvenating old satellite cells.Citation46,47 Growth differentiation factor 11 (GDF11) and Oxytocin are systemic factors depleted in the serum from old animals, and its supplementation was sufficient to restore muscle regeneration capacity and improve myogenic potential and genomic integrity in old satellite cells.Citation46,47 Of note, GDF11 is a TGFβ-family member, as Myostatin,Citation48 and given the anti-myogenic and pro-atrophying roles of this growth factor family, the pro-regenerative function of GFD11 came as unexpected.
Together, these studies support the idea that there is in fact an important contribution of the environment to the process of satellite cell aging, and that many of the changes that occur during muscle stem cell aging can be reversed. While this model holds true for classically classified old animals (around 20 months of age), one would expect that if age-associated senescence is a consequence of combined cellular stresses as recently proposed,Citation25 they would take a longer period of time to accumulate and senescent cells may not be observed until later stages in life. Here, we present and discuss our recent dataCitation49 showing that in “healthy” geriatric animals (beyond 28 months), satellite cells enter an irreversible pre-senescent state that affects their intrinsic regenerative and self-renewal ability. We propose that maintenance of quiescence in adult life depends on the active repression of senescent pathways and that the molecular mechanisms behind satellite cell geroconversion in geriatric animals relay on the de-repression of the p16INK4a locus. Because senescent cells have also been observed in patients with several age-related pathologies, associated with the affected tissues,Citation24,50,51 including cancer, which also increases with age,Citation52 we propose the geroconversion into a senescent state might be common to stem cells in different tissues and the underlying cause of several age-related dysfunctions.
Old vs Geriatric Satellite Cells: An Irreversible Switch
Although old animals show features consistent with the onset of muscle atrophy it is not until geriatric ages that a full sarcopenic phenotype can be detected. Consistent with this, regenerative capacity after muscle injury, which is already decreased in old animals is further impaired in geriatric animals, correlating with a progressive decrease in activation rate of satellite cells in response to injury. Interestingly, the worsening of the phenotype is not due to a loss of satellite cells. Satellite cell numbers decrease in old animals compared to adult (5-6 months old) mice,Citation32,53 but this decline in number is no further accentuated after 24 months of age.Citation49 This observation suggested to us that there may be intrinsic changes that occur in geriatric satellite cells that underlie the marked drop in regenerative ability and satellite cell activation potential observed in geriatric animals. To address this question, we have isolated satellite cells from donor mice of different ages—adult, old and geriatric—and compared their regenerative and self-renewal capabilities. Consistent with previous studies (see above), satellite cells isolated from old mice and transplanted into injured muscles of young hosts were able to regenerate the muscle with an efficiency similar to adult satellite cells. In agreement, old satellite cell activation potential was not significantly distinct to that of adult satellite cells after transplant into young hosts. These observations confirm that old satellite cells can be rejuvenated by exposure to a young environment and their loss of regenerative ability and activation potential can be rescued. Importantly, satellite cells isolated from geriatric mice and transplanted into injured muscles of young hosts remained unable to regenerate the damaged muscle, indicating that the impairment in geriatric satellite cell function cannot be reversed by exposure to a youthful environment.Citation49 Supporting this idea is the observation that geriatric satellite cell activation remained impaired after transplant into young hosts, even after a 21 d adaptation period. These observations strongly suggested that there were intrinsic changes that occur specifically in geriatric satellite cells that cannot be reversed by youthful environmental cues.
We have used global gene expression analysis of satellite cells freshly isolated from resting skeletal muscle to identify transcriptional programs that may be responsible for the intrinsic changes described above.Citation54 Interestingly, a gene set enrichment analysis (GSEA)Citation55 of the significantly upregulated genes in geriatric satellite cells was enriched in gene sets controlled by Polycomb repressive complexes (PRC1 and PRC2), which are known mediators of cellular senescence.Citation56 Furthermore, comparison with a comprehensive list of genes implicated in cellular senescenceCitation57-59 showed specific up-regulation of several genes, including the master regulator of senescence Cdkn2a (p16INK4a).Citation15,17,49,60,61
Satellite Cell Geroconversion
The switch into a pre-senescent state anticipated by the transcriptome profile led to the hypothesis that in geriatric animals a quiescent to senescent conversion in muscle stem cells may be the underlying cause for an irreversible impairment in activation/proliferative potential, required both for self-renewal and for the proliferative expansion that precedes myogenesis. Indeed, we observed that after successive rounds of muscle injury, geriatric but not old satellite cells showed a progressive and cumulative loss of self-renewal ability. Consistently, the proliferative capacity of geriatric satellite cells induced by regeneration in vivo or growth conditions in vitro was markedly reduced. Confirming the switch from a quiescent to a pre-senescent state, satellite cells from geriatric animals placed under this proliferative pressure acquired characteristics of full senescence, such as senescence-associated β-galactosidase activity and expression of several senescence markers. These features were observed both in vivo and in vitro and could not be reversed by adaptation to a youthful environment confirming that the quiescence-to-senescence switch of geriatric satellite cells is irreversible and leads to accelerated senescence (i.e., geroconversion). Interestingly, these features of cellular senescence were also observed in human satellite cells from sarcopenic geriatric individuals, which express p16INK4a and other senescence markers. This is the first time that senescent stem cells are observed in vivo during skeletal muscle's normal aging and supports the previously proposed hypothesis that senescence may indeed be a driving force for age-associated tissue dysfunction and regenerative decline.Citation25,26,49
p16INK4a/Rb/E2F Axis in Satellite Cell Geroconversion
INK4a refers to a chromosomal locus encoding for 2 alternative reading frame proteins described as tumor suppressors: p16INK4a and ARF (p19ARF in mouse and p14ARF in human).Citation62 Each of these proteins has been shown to have a critical role in senescence, cell cycle control and tumor suppression, and this locus is often functionally inactivated in human tumors. p16INK4a is a cyclin D-dependent kinase (cdk) inhibitor acting upstream of retinoblastoma protein (pRb), preventing its phosphorylation and functional inactivation by cdks.Citation63
p16INK4a gene expression increases with age in a variety of tissuesCitation64 and induction of p16INK4a expression can cause senescence of a variety of cell types in culture and in vivo.Citation63 Although a universal marker that is exclusively expressed in senescent cells has not been identified, most senescent cells seem to express p16INK4a, making it a generally accepted biomarker of senescence. Removal of senescent cells using a strategy of inducible elimination of p16INK4a-expressing cells upon drug administration delays the onset of some of the most common age-related phenotypes in adipose tissue, skeletal muscle and eye of BubR1 progeroid mice,Citation17 reinforcing the idea that p16INK4a associated senescence plays a central role in age-dependent tissue dysfunction.
Because p16INK4a was the only senescence-associated gene identified by our global gene expression profile to be up-regulated in different sarcopenia-associated satellite cell signatures, we sought to investigate a role for p16INK4a in satellite cell geroconversion. p16INK4a overexpression in young satellite cells was sufficient to prevent efficient activation and to promote geroconversion under proliferative pressure both during regeneration and in vitro growth conditions, suggesting that p16INK4a is a central molecular effector of quiescence-to-senescence switch of satellite cells (). Because p16INK4a acts upstream of retinoblastoma/E2F mediated repression of proliferation-promoting transcription factors we sought to investigate the involvement of this pathway in satellite cell geroconversion. Indeed, geriatric satellite cells which express high levels of p16INK4a under proliferative pressure also show a reduction in the phosphorylated form of retinoblastoma and a consequent repression of Rb/E2F target genes required for cell cycle progression. Importantly, the activation of this Rb/E2F axis could not be reversed by environmental cues, when geriatric satellite cells were transplanted into injured young muscles. Consistent with the senescent phenotype observed in human geriatric satellite cells, Rb phosphorylation and Rb/E2F target genes are also repressed in satellite cells from geriatric sarcopenic individuals.Citation49 In agreement with our findings, a coetaneous publication of the Blau lab,Citation65 reported that the activation of the p38 MAPK cascade in aged satellite cells, as it has been demonstrated in different somatic cell types,Citation66-70 could underlie p16INK4a, and that inhibition of the p38 MAPK pathway rejuvenated the satellite cells.Citation65
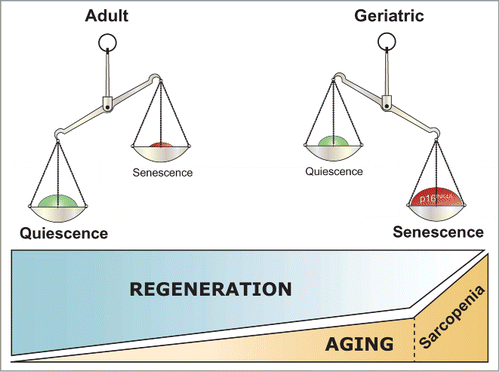
Figure 1. Satellite cell geroconversion disrupts muscle regeneration. Satellite cells stay quiescent in young and adult mice under normal conditions. During aging, geriatric satellite cells of sarcopenic muscles undergo derepression of p16INK4a, a regulator of cellular senescence, losing as a consequence their reversible quiescent state. Instead, they adopt a senescent-like state (becoming pre-senescent cells), which impairs the regeneration process by specifically preventing stem cell activation, proliferation and self-renewal.
Epigenetic Regulation of the p16INK4a Locus
Given the central role of the p16INK4a on satellite cell geroconversion, it is important to understand the mechanisms involved in the regulation of its locus in the context of satellite cell aging. Epigenetic regulation through Polycomb repressive complexes is a key component of p16INK4a silencing and is achieved through the sequential establishment of the H3K27me3 chromatin mark and the mono-ubiquitination of histone H2A, by PRC2 and PRC1 complexes, respectively.Citation71-73 Bmi1 (a member of the PRC1 complex) is found strongly enriched at the INK4a promoter, together with PRC2 members and the associated H3K27me3 mark, in different tissues.Citation74-76 The critical role of PRCs in regulating the INK4A locus is further supported by studies showing that over-expression of PRC1 and PRC2 components is sufficient to bypass senescence, while cells lacking PRC1 components, including Bmi1, show aberrant expression of the p16INK4a gene.Citation15,75,77 In agreement with the extensive data on the regulatory functions of PRC proteins, we found that also in young satellite cells the INK4a locus (both at the RD domain and at exon1a/2 regions,Citation78,79 was marked with the PRC1 repressive mark H2AUb and that this mark was significantly reduced in satellite cells from geriatric mice.Citation49 This pattern of locus occupancy is consistent with p16INK4a transcriptional repression and induction in young and geriatric satellite cells, respectively.
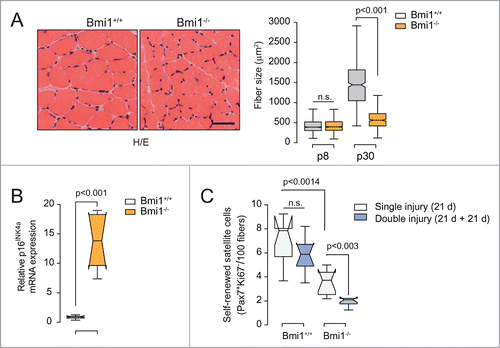
To explore the relevance of INK4a locus derepression and associated p16INKa expression in satellite cell function, we used Bmi1-deficient mice. Bmi1−/− mice display a number of defects including a progressive decrease in the number of haematopoietic cells, severe neurological abnormalities and markedly shortened lifespan.Citation80 Young adult Bmi1-deficient mice were considerably smaller than the age-matched WT littermates and displayed a corresponding reduction in muscle size (). Evaluation of myofiber cross-sectional area showed that these differences were not significant at early post-natal stages but dramatically increased thereafter, suggesting premature muscle wasting (), and in agreement with a previous report.Citation81 Several studies indicate that Bmi1 is required for the self-renewal and post-natal maintenance of the stem cell population of haematopoietic, neural, gastrointestinal and breast tissues.Citation82-85 In most cases, the defect in self-renewal in the absence of Bmi1 was associated with increased p16INK4a expression and INK4a deficiency partially rescues haematopoietic and neural stem cell self-renewal of Bmi1−/− mice.Citation86,87 We reasoned that loss of repression of the INK4a locus and the high expression of p16INK4a might also provoke defective self-renewal in Bmi1 null satellite cells (). We forced these cell to undergo cycles of self-renewal through sequential rounds of quiescence, activation, proliferation and return to quiescence, by performing successive rounds of muscle injury. We quantified self-renewal efficiency by the number of quiescent (Pax7+/Ki67−) satellite cells associated to newly formed myofibers. 21 days after CTX injury, the number of Pax7+/Ki67− satellite cells was significantly reduced in Bmi1−/− regenerated fibers compared to the WT (). The regenerated muscle was re-injured to trigger the reactivation of satellite cells from the quiescent state, new round of myogenesis and self-renewal. Importantly, the number of self-renewed satellite cells was further reduced in Bmi1-deficient mice correlating with a regeneration defect and an upregulation of p16INK4a (). These results indicate that a deregulation of the Bmi1/p16INK4a axis leads to an intrinsic defect in satellite cell self-renewal, that mimics what is observed in geriatric animals.Citation49
Figure 2. Atrophic muscles and pre-senescent satellite cells in Bmi1-deficient mice. (A) Right: Representative images of Hematoxilin/Eosin (H/E) stained cryosections of tibialis anterior (TA) muscle from Bmi1+/+ and Bm1−/− animals at 30 d after birth (p30). Left: Boxplot represent the quantification of myofiber size, evaluated by the cross-sectional area at the indicated days after birth. n = 10 mice, 500 fibers were measured per mice. (B) Expression of p16INK4a, evaluated by RT-qPCR, in satellite cells isolated from Bmi1+/+ and Bm1−/− mice by FACS as described.Citation49 n = 7 mice. (C) Self-renewed satellite cells (Pax7+/Ki67−) identified by immunostaining in cryosections from regenerating TA muscle of Bmi1+/+ and Bm1−/− mice analyzed 21 d after being injury-induced by cardiotoxin injection (single muscle injury). Twenty one days after the first injury, the muscles were re-injured and satellite cell self-renewal was analyzed 21 d after the second injury (double muscle injury). n = 7 mice. Scale bars, 50 μm. Data are mean ± s.e.m. Two-sided Mann-Whitney U test was used to assess statistical significance. P values are indicated. n.s. not significant. Boxplot center lines show the medians; box limits indicate the 25th and 75th percentiles as determined by R software;Citation93 whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles.
Genetic Rejuvenation of Geriatric Satellite Cells
Our data suggest that there is an active protective mechanism repressing senescence pathways in young satellite cells, and that its deregulation in geriatric individuals leads to intrinsic changes to the satellite cells that cannot be reversed by a youthful environment. However, this does not mean that senescence cannot be overcome and geriatric satellite cells rejuvenated to restore muscle regeneration in sarcopenic individuals. On the contrary, our results show that p16INK4a inhibition is an effective rejuvenation strategy for geriatric satellite cells. p16INK4a silencing with short hairpin specifically in isolated geriatric satellite cells before transplantation into young muscles was sufficient to restore activation potential. p16INK4a silencing also restored self-renewal capacity of satellite cells both in vivo and in vitro and improved regeneration efficiency. Importantly, both in mouse and human satellite cells, p16INK4a silencing alone limited satellite cell geroconversion, as shown by enhanced proliferative ability and reduced senescence makers.Citation49 This suggests that genetic rejuvenation of satellite cells by preventing p16INK4a expression may be an effective way to prevent poor regeneration phenotypes in humans undergoing sarcopenia, though some concerns apply, first whether such therapeutic intervention will be feasible and second whether the benefits of a muscle stem cell rejuvenation outweigh the possible risks of a reduction of a tumor-suppressor in the context of aging.
Understanding how intrinsic aging is triggered may be the key to bypass these concerns. In that sense, as mentioned above, Cosgrove et al. have recently demonstrated that the p38 MAPK cascade is part of the signaling pathway leading to p16INK4a expression and that its pharmacological inhibition is able to rescue the poor proliferation phenotype of old satellite cells.Citation65 The environmental or niche extrinsic factors that may be implicated in the activation of the p38 MAPK pathway remain mostly unknown, though it has been shown that FGF-2 signals may be upstream of p38 MAPK pathway in the committed cell during asymmetric division of young satellite cells,Citation88 and that in an aging environment FGF-2 levels increase, asymmetric division is impaired and symmetric division with p38 MAPK activation in both cells is induced.Citation89 Inhibition of the p38 MAPK pathway restores asymmetric division and aged satellite cell function.Citation89 Though inhibition of p38 MAPK is increasingly considered as a therapeutic strategy -mostly because of its role as a master regulator of inflammatory responses-, the facts that p38 MAPKs are crucial stress-responsive kinases expressed in every cell and the discovery of p38 MAPK as a main actor in the development of tumorigenesisCitation90 have generated many concerns over its practical use in long term therapies. While this paper was being reviewed, two new papers reported the involvement of the JAK/STAT signaling pathway in the reduced expansion of satellite cells with aging, and the rescue of this defect by JAK/STAT inhibitors.Citation91,92
Concluding Remarks
In vivo stem cell senescence is an emerging concept that may help understand and design strategies to treat several age related pathologies. Our data shows that the maintenance of satellite cell quiescence throughout adult life requires the active repression of genes associated with senescence and that the loss of epigenetic marks regulating these genes in advanced aging results in a quiescence-to-senescence cellular switch. Because the pathways involved in cellular senescence are conserved, it is likely that cellular senescence occurs in other tissues during physiological aging and that this may be a common mechanism that coordinates tissue aging at the organism level. Understanding the underlying mechanism involved in the de-repression of these pathways with advanced aging, and particularly in aging stem cells, may help design strategies to delay the aging process and the age-associated tissue regenerative decline. However, because senescence has been shown to have other physiological roles beyond aging, strategies involving the repression of senescence pathways should be looked at with care and be considered in an age- and cell/tissue-dependent manner.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are indebted to AL Serrano, L García-Prat, S Gutarra, L Ortet and members of the Cell Biology Group for their contributions to this study.
Funding
The authors acknowledge funding from MINECO-Spain (SAF2012–38547, FIS-PI13/025, PLE2009–0124), DuchennePP-NL, E-Rare, Fundació Marató TV3, MDA, EU-FP7 (Myoage, Optistem and Endostem) and AFM.
References
- Rando TA. Stem cells, ageing and the quest for immortality. Nature 2006; 441:1080-6; PMID:16810243; http://dx.doi.org/10.1038/nature04958
- Jung Y, Brack AS. Cellular mechanisms of somatic stem cell aging. Curr Top Dev Biol 2014; 107:405-38; PMID:24439814; http://dx.doi.org/10.1016/B978-0-12-416022-4.00014-7
- Motohashi N, Asakura A. Muscle satellite cell heterogeneity and self-renewal. Front Cell Dev Biol 2014; 2; PMID:24977132; http://dx.doi.org/10.3389/fcell.2014.00001
- Muller-Sieburg CE, Sieburg HB, Bernitz JM, Cattarossi G. Stem cell heterogeneity: implications for aging and regenerative medicine. Blood 2012; 119:3900-7; PMID:22408258; http://dx.doi.org/10.1182/blood-2011-12-376749
- Liu L, Rando TA. Manifestations and mechanisms of stem cell aging. J Cell Biol 2011; 193:257-66; PMID:21502357; http://dx.doi.org/10.1083/jcb.201010131
- Li L, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science 2010; 327:542-5; PMID:20110496; http://dx.doi.org/10.1126/science.1180794
- Garcia-Prat L, Sousa-Victor P, Munoz-Canoves P. Functional dysregulation of stem cells during aging: a focus on skeletal muscle stem cells. FEBS J 2013; 280:4051-62; PMID:23452120; http://dx.doi.org/10.1111/febs.12221
- Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Nat Acad Sci U S A 2012; 109:13314-8; PMID:22847439; http://dx.doi.org/10.1073/pnas.1205690109
- Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging 2012; 4:159-65; PMID:22394614
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961; 25:585-621; PMID:13905658; http://dx.doi.org/10.1016/0014-4827(61)90192-6
- Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006; 443:453-7; PMID:16957737; http://dx.doi.org/10.1038/nature05092
- Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev 2007; 128:36-44; PMID:17116315; http://dx.doi.org/10.1016/j.mad.2006.11.008
- Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009; 8:311-23; PMID:19627270; http://dx.doi.org/10.1111/j.1474-9726.2009.00481.x
- Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell 2004; 14:501-13; PMID:15149599; http://dx.doi.org/10.1016/S1097-2765(04)00256-4
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006; 443:448-52; PMID:16957738; http://dx.doi.org/10.1038/nature05091
- Baker DJ, Perez-Terzic C, Jin F, Pitel KS, Niederlander NJ, Jeganathan K, Yamada S, Reyes S, Rowe L, Hiddinga HJ, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol 2008; 10:825-36; PMID:18516091; http://dx.doi.org/10.1038/ncb1744
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011; 479:232-6; PMID:22048312; http://dx.doi.org/10.1038/nature10600
- Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, Nasto LA, St Croix CM, Usas A, Vo N, et al. NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest 2012; 122:2601-12; PMID:22706308; http://dx.doi.org/10.1172/JCI45785
- Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013; 155:1119-30; PMID:24238961; http://dx.doi.org/10.1016/j.cell.2013.10.041
- Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, et al. Programmed cell senescence during mammalian embryonic development. Cell 2013; 155:1104-18; PMID:24238962; http://dx.doi.org/10.1016/j.cell.2013.10.019
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell 2008; 134:657-67; PMID:18724938; http://dx.doi.org/10.1016/j.cell.2008.06.049
- Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 2010; 12:676-85; PMID:20526329; http://dx.doi.org/10.1038/ncb2070
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88:593-602; PMID:9054499; http://dx.doi.org/10.1016/S0092-8674(00)81902-9
- Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014; 15:482-96; PMID:24954210; http://dx.doi.org/10.1038/nrm3823
- van Deursen JM. The role of senescent cells in ageing. Nature 2014; 509:439-46; PMID:24848057
- Baker DJ, Weaver RL, van Deursen JM. p21 both attenuates and drives senescence and aging in BubR1 progeroid mice. Cell Rep 2013; 3:1164-74; PMID:23602569; http://dx.doi.org/10.1016/j.celrep.2013.03.028
- Gros J, Manceau M, Thome V, Marcelle C. A common somitic origin for embryonic muscle progenitors and satellite cells. Nature 2005; 435:954-8; PMID:15843802; http://dx.doi.org/10.1038/nature03572
- Kassar-Duchossoy L, Giacone E, Gayraud-Morel B, Jory A, Gomes D, Tajbakhsh S. Pax3/Pax7 mark a novel population of primitive myogenic cells during development. Genes Dev 2005; 19:1426-31; PMID:15964993; http://dx.doi.org/10.1101/gad.345505
- Relaix F, Montarras D, Zaffran S, Gayraud-Morel B, Rocancourt D, Tajbakhsh S, Mansouri A, Cumano A, Buckingham M. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J Cell Biol 2006; 172:91-102; PMID:16380438; http://dx.doi.org/10.1083/jcb.200508044
- Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev 2013; 93:23-67; PMID:23303905; http://dx.doi.org/10.1152/physrev.00043.2011
- Hikida RS. Aging changes in satellite cells and their functions. Curr Aging Sci 2011; 4:279-97; PMID:21529324; http://dx.doi.org/10.2174/1874609811104030279
- Chakkalakal JV, Jones KM, Basson MA, Brack AS. The aged niche disrupts muscle stem cell quiescence. Nature 2012; 490:355-60; PMID:23023126; http://dx.doi.org/10.1038/nature11438
- Zwetsloot KA, Childs TE, Gilpin LT, Booth FW. Non-passaged muscle precursor cells from 32-month old rat skeletal muscle have delayed proliferation and differentiation. Cell Proliferat 2013; 46:45-57; PMID:23279018; http://dx.doi.org/10.1111/cpr.12007
- Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, Rando TA. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 2007; 317:807-10; PMID:17690295; http://dx.doi.org/10.1126/science.1144090
- Shefer G, Wleklinski-Lee M, Yablonka-Reuveni Z. Skeletal muscle satellite cells can spontaneously enter an alternative mesenchymal pathway. J Cell Sci 2004; 117:5393-404; PMID:15466890; http://dx.doi.org/10.1242/jcs.01419
- Taylor-Jones JM, McGehee RE, Rando TA, Lecka-Czernik B, Lipschitz DA, Peterson CA. Activation of an adipogenic program in adult myoblasts with age. Mech Ageing Dev 2002; 123:649-61; PMID:11850028; http://dx.doi.org/10.1016/S0047-6374(01)00411-0
- Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011; 477:90-4; PMID:21886162; http://dx.doi.org/10.1038/nature10357
- Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 2005; 433:760-4; PMID:15716955; http://dx.doi.org/10.1038/nature03260
- Gutmann E, Carlson BM. Regeneration and transplantation of muscles in old rats and between young and old rats. Life Sci 1976; 18:109-14; PMID:1250056; http://dx.doi.org/10.1016/0024-3205(76)90280-0
- Harrison DE. Long-term erythropoietic repopulating ability of old, young, and fetal stem cells. J Exp Med 1983; 157:1496-504; PMID:6854204; http://dx.doi.org/10.1084/jem.157.5.1496
- Carlson BM, Faulkner JA. The regeneration of skeletal muscle fibers following injury: a review. MedSciSports Exerc 1983; 15:187.
- Carlson BM, Faulkner JA. Muscle transplantation between young and old rats: age of host determines recovery. Am J Physiol 1989; 256:C1262-6; PMID:2735398
- Roberts P, McGeachie JK, Grounds MD. The host environment determines strain-specific differences in the timing of skeletal muscle regeneration: cross-transplantation studies between SJL/J and BALB/c mice. J Anat 1997; 191 (Pt 4):585-94; PMID:9449077; http://dx.doi.org/10.1046/j.1469-7580.1997.19140585.x
- Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 2008; 454:528-32; PMID:18552838; http://dx.doi.org/10.1038/nature07034
- Shea KL, Xiang W, LaPorta VS, Licht JD, Keller C, Basson MA, Brack AS. Sprouty1 regulates reversible quiescence of a self-renewing adult muscle stem cell pool during regeneration. Cell Stem Cell 2010; 6:117-29; PMID:20144785; http://dx.doi.org/10.1016/j.stem.2009.12.015
- Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R, Miller C, Regalado SG, Loffredo FS, Pancoast JR, et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science 2014; 344:649-52; PMID:24797481; http://dx.doi.org/10.1126/science.1251152
- Elabd C, Cousin W, Upadhyayula P, Chen RY, Chooljian MS, Li J, Kung S, Jiang KP, Conboy IM. Oxytocin is an age-specific circulating hormone that is necessary for muscle maintenance and regeneration. Nat Commun 2014; 5:4082; PMID:24915299; http://dx.doi.org/10.1038/ncomms5082
- Lee YS, Lee SJ. Regulation of GDF-11 and myostatin activity by GASP-1 and GASP-2. Proc Nat Acad Sci U S A 2013; 110:E3713-22; PMID:24019467; http://dx.doi.org/10.1073/pnas.1309907110
- Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardi M, Ballestar E, Gonzalez S, Serrano AL, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014; 506:316-21; PMID:24522534; http://dx.doi.org/10.1038/nature13013
- Campisi J. Aging, cellular senescence, and cancer. Ann Rev Physiol 2013; 75:685-705; PMID:23140366; http://dx.doi.org/10.1146/annurev-physiol-030212-183653
- Naylor RM, Baker DJ, van Deursen JM. Senescent cells: a novel therapeutic target for aging and age-related diseases. Clin Pharmacol Therap 2013; 93:105-16; PMID:23212104; http://dx.doi.org/10.1038/clpt.2012.193
- Blagosklonny MV. Selective anti-cancer agents as anti-aging drugs. Cancer Biol Ther 2013; 14:1092-7; PMID:24345884; http://dx.doi.org/10.4161/cbt.27350
- Shefer G, Van de Mark DP, Richardson JB, Yablonka-Reuveni Z. Satellite-cell pool size does matter: defining the myogenic potency of aging skeletal muscle. Dev Biol 2006; 294:50-66; PMID:16554047; http://dx.doi.org/10.1016/j.ydbio.2006.02.022
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57; PMID:19131956; http://dx.doi.org/10.1038/nprot.2008.211
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Nat Acad Sci U S A 2005; 102:15545-50; PMID:16199517; http://dx.doi.org/10.1073/pnas.0506580102
- Lanigan F, Geraghty JG, Bracken AP. Transcriptional regulation of cellular senescence. Oncogene 2011; 30:2901-11; PMID:21383691; http://dx.doi.org/10.1038/onc.2011.34
- Fridman AL, Tainsky MA. Critical pathways in cellular senescence and immortalization revealed by gene expression profiling. Oncogene 2008; 27:5975-87; PMID:18711403; http://dx.doi.org/10.1038/onc.2008.213
- Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, et al. Tumour biology: senescence in premalignant tumours. Nature 2005; 436:642; PMID:16079833; http://dx.doi.org/10.1038/436642a
- Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Ann Rev Pathol 2010; 5:99-118; PMID:20078217; http://dx.doi.org/10.1146/annurev-pathol-121808-102144
- Jacobs JJ, de Lange T. Significant role for p16INK4a in p53-independent telomere-directed senescence. Curr Biol: CB 2004; 14:2302-8; PMID:15620660; http://dx.doi.org/10.1016/j.cub.2004.12.025
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 2006; 443:421-6; PMID:16957735
- Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol 2001; 2:731-7; PMID:11584300; http://dx.doi.org/10.1038/35096061
- Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev 2003; 13:77-83; PMID:12573439; http://dx.doi.org/10.1016/S0959-437X(02)00013-8
- Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest 2004; 114:1299-307; PMID:15520862; http://dx.doi.org/10.1172/JCI22475
- Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL, Blau HM. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med 2014; 20:255-64; PMID:24531378; http://dx.doi.org/10.1038/nm.3464
- Wong ES, Le Guezennec X, Demidov ON, Marshall NT, Wang ST, Krishnamurthy J, Sharpless NE, Dunn NR, Bulavin DV. p38MAPK controls expression of multiple cell cycle inhibitors and islet proliferation with advancing age. Dev Cell 2009; 17:142-9; PMID:19619499; http://dx.doi.org/10.1016/j.devcel.2009.05.009
- Zhang Y, Herbert BS, Rajashekhar G, Ingram DA, Yoder MC, Clauss M, Rehman J. Premature senescence of highly proliferative endothelial progenitor cells is induced by tumor necrosis factor-alpha via the p38 mitogen-activated protein kinase pathway. FASEB J: Off Publ Fed Am Soc Exp Biol 2009; 23:1358-65; PMID:19124561; http://dx.doi.org/10.1096/fj.08-110296
- Iwasa H, Han J, Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells: Dev Mol Cell Mech 2003; 8:131-44; http://dx.doi.org/10.1046/j.1365-2443.2003.00620.x
- Zheng H, Seit-Nebi A, Han X, Aslanian A, Tat J, Liao R, Yates JR 3rd, Sun P. A posttranslational modification cascade involving p38, Tip60, and PRAK mediates oncogene-induced senescence. Mol Cell 2013; 50:699-710; PMID:23685072; http://dx.doi.org/10.1016/j.molcel.2013.04.013
- Lanna A, Henson SM, Escors D, Akbar AN. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat Immunol 2014; 15:965-72; PMID:25151490; http://dx.doi.org/10.1038/ni.2981
- Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004; 431:873-8; PMID:15386022; http://dx.doi.org/10.1038/nature02985
- Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell 2005; 20:845-54; PMID:16359901; http://dx.doi.org/10.1016/j.molcel.2005.12.002
- Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 2009; 10:697-708; PMID:19738629; http://dx.doi.org/10.1038/nrm2763
- Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999; 397:164-8; PMID:9923679; http://dx.doi.org/10.1038/16476
- Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, Theilgaard-Monch K, Minucci S, Porse BT, Marine JC, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev 2007; 21:525-30; PMID:17344414; http://dx.doi.org/10.1101/gad.415507
- Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 2008; 32:503-18; PMID:19026781; http://dx.doi.org/10.1016/j.molcel.2008.11.004
- Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol 2006; 7:667-77; PMID:16921403; http://dx.doi.org/10.1038/nrm1987
- Agherbi H, Gaussmann-Wenger A, Verthuy C, Chasson L, Serrano M, Djabali M. Polycomb mediated epigenetic silencing and replication timing at the INK4a/ARF locus during senescence. PLoS One 2009; 4:e5622; PMID:19462008; http://dx.doi.org/10.1371/journal.pone.0005622
- Gonzalez S, Klatt P, Delgado S, Conde E, Lopez-Rios F, Sanchez-Cespedes M, Mendez J, Antequera F, Serrano M. Oncogenic activity of Cdc6 through repression of the INK4/ARF locus. Nature 2006; 440:702-6; PMID:16572177; http://dx.doi.org/10.1038/nature04585
- van der Lugt NM, Domen J, Linders K, van Roon M, Robanus-Maandag E, te Riele H, van der Valk M, Deschamps J, Sofroniew M, van Lohuizen M, et al. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev 1994; 8:757-69; PMID:7926765; http://dx.doi.org/10.1101/gad.8.7.757
- Robson LG, Di Foggia V, Radunovic A, Bird K, Zhang X, Marino S. Bmi1 is expressed in postnatal myogenic satellite cells, controls their maintenance and plays an essential role in repeated muscle regeneration. PLoS One 2011; 6:e27116; PMID:22096526; http://dx.doi.org/10.1371/journal.pone.0027116
- Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res 2006; 66:6063-71; PMID:16778178; http://dx.doi.org/10.1158/0008-5472.CAN-06-0054
- Tateishi K, Ohta M, Kanai F, Guleng B, Tanaka Y, Asaoka Y, Tada M, Seto M, Jazag A, Lianjie L, et al. Dysregulated expression of stem cell factor Bmi1 in precancerous lesions of the gastrointestinal tract. Clin Cancer Res 2006; 12:6960-6; PMID:17145814; http://dx.doi.org/10.1158/1078-0432.CCR-06-0449
- Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 2003; 425:962-7; PMID:14574365; http://dx.doi.org/10.1038/nature02060
- Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003; 423:255-60; PMID:12714970; http://dx.doi.org/10.1038/nature01572
- Smith KS, Chanda SK, Lingbeek M, Ross DT, Botstein D, van Lohuizen M, Cleary ML. Bmi-1 regulation of INK4A-ARF is a downstream requirement for transformation of hematopoietic progenitors by E2a-Pbx1. Mol Cell 2003; 12:393-400; PMID:14536079; http://dx.doi.org/10.1016/S1097-2765(03)00277-6
- Molofsky AV, He S, Bydon M, Morrison SJ, Pardal R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev 2005; 19:1432-7; PMID:15964994; http://dx.doi.org/10.1101/gad.1299505
- Jones NC, Tyner KJ, Nibarger L, Stanley HM, Cornelison DD, Fedorov YV, Olwin BB. The p38alpha/beta MAPK functions as a molecular switch to activate the quiescent satellite cell. J Cell Biol 2005; 169:105-16; PMID:15824134; http://dx.doi.org/10.1083/jcb.200408066
- Bernet JD, Doles JD, Hall JK, Kelly Tanaka K, Carter TA, Olwin BB. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat Med 2014; 20:265-71; PMID:24531379; http://dx.doi.org/10.1038/nm.3465
- Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 2009; 9:537-49; PMID:19629069; http://dx.doi.org/10.1038/nrc2694
- Tierney MT, Aydogdu T, Sala D, Malecova B, Gatto S, Puri PL, Latella L, Sacco A. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat Med 2014.
- Price FD, von Maltzahn J, Bentzinger CF, Dumont NA, Yin H, Chang NC, Wilson DH, Frenette J, Rudnicki MA. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat Med 2014.
- Spitzer M, Wildenhain J, Rappsilber J, Tyers M. BoxPlotR: a web tool for generation of box plots. Nat Methods 2014; 11:121-2; PMID:24481215; http://dx.doi.org/10.1038/nmeth.2811