
Abstract
Random amplified polymorphic DNA (RAPD) analysis was adapted for genomic identification of cell cultures and evaluation of DNA stability in cells of different origin at different culture passages. DNA stability was observed in cultures after no more than 5 passages. Adipose-derived stromal cells demonstrated increased DNA instability. RAPD fragments from different cell lines after different number of passages were cloned and sequenced. The chromosomal localization of these fragments was identified and single-nucleotide variations in RAPD fragments isolated from cell lines after 8–12 passages were revealed. Some of them had permanent localization, while most variations demonstrated random distribution and can be considered as de novo mutations.
Introduction
The application of cell therapy brings up the problem of the safety of transplanted cells. At the same time, cultivation of cell material can introduce modifications to the genomic DNA dangerous for the patient. Karyotype analysis of cell lines and cultures demonstrated chromosomal abnormalities and a trend to karyotype instability.Citation1-3 In addition, cell growth rate as well as their differentiation potential can significantly change upon culturing.Citation4-6 In this context, culture and transfection of human cell lines require stringent control of genomic alterations in transplanted cells. We assumed that RAPD analysis can be used for this purpose. The diagnostic potential of RAPD technique has been successfully demonstrated for the description of genetic variation of microorganisms, higher plants, invertebrates, and vertebrates.Citation7-10 The most detailed RAPD analysis was performed for crops, livestock, and laboratory animals for the identification and differentiation of breeds and individual lines, chromosomal mapping, and identification of commercially valuable characters.Citation7-10 The studies of Dil-AfrozeCitation11 and OngCitation12 demonstrated the applicability of RAPD analysis for the detection of genetic instability in brain and lung cancer. They proposed RAPD analysis as an additional test for genomic rearrangements in cancer. Later this approach was used to study genomic instability in liver cancer in transgenic miceCitation13 and human hepatocellular carcinoma.Citation14 RAPD was also applied to detect somatic alterations in azoxymethane-induced rat colon tumors.Citation15
Thus, RAPD analysis is an efficient tool for the identification of DNA alterations in malignant cells; however, the data on DNA variation in normal cells, particularly, after long-term culture are clearly insufficient. Long-term culture-induced genetic abnormalities initially include occasional point mutations, which can substantially affect genetic control. The goal of this work was to study genomic instability in human cell cultures of different origin using RAPD analysis and to identify the types of structural variations in DNA amplified from different passages.
Results
RAPD analysis of cell cultures
Structural DNA variations in human cells at different culture stages were analyzed by RAPD using PCR primers previously approved in our study of somatic mosaicism in humans.Citation16 Zero passage refers to the culture before medium replacement; first passage, to the first subculture. The results of DNA amplification with the P29 primer are shown in .
Figure 1. RAPD analysis of DNA from cell cultures at different passages using the P29 primer. A 100 bp Ladder+ and a 1 kb Ladder (Fermentas) were used as molecular weight markers. Culture nos. 1–5, adipose-derived stromal cells; MSCs, mesenchymal stem cells; nos. 11, 14, 16, 18–21, and 23, skin fibroblasts; lanes 1–20, different passages; ‘*’ indicates that the cell line was cultured on different medium; nos.1–23, cell cultures obtained from different donors.
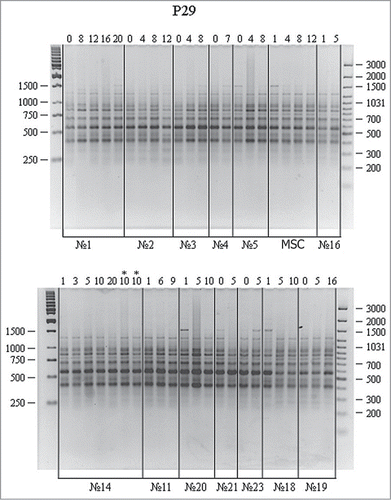
An average RAPD spectra included 8 fragments in the range of 300 to 1500 bp. RAPD spectra proved identical for the vast majority of cultures (nos. 1–23). The differences between certain spectra (for instance, lane 1 in culture no. 18, lane 1 in culture no. 20, and lane 1 in MSCs) have not been confirmed in subsequent experiments and were ignored. No differences between RAPD spectra of different passages in the same cell culture have been revealed either. Again, minor variations have not been confirmed in subsequent experiments. It should be specifically noted that medium replacement had no effect on RAPD spectra (lanes 10 and 10* – culturing 10 using another culture medium in culture no. 14). Thus, no significant differences in RAPD spectra have been revealed for the studied DNA samples using the P29 primer.
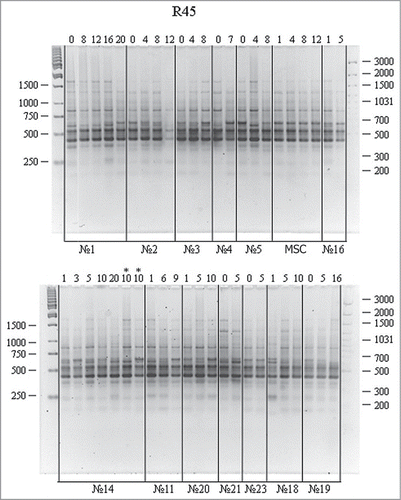
RAPD analysis of the same DNA samples using the R45 primer is shown in . The amplification spectra had ∼8 fragments (including 3–4 major ones) in the range of 300 to 1500 bp. Most spectra were similar for different passages. Minor distinctions including band shift, doubling, or disappearance in the region of 600 bp were observed. Subsequent experiments reproduced these variations. Minor band variations in the region above 1000 bp were not reproduced in subsequent experiments and were ignored.
Figure 2. RAPD analysis of DNA from cell cultures at different passages using the R45 primer. For designations, see .
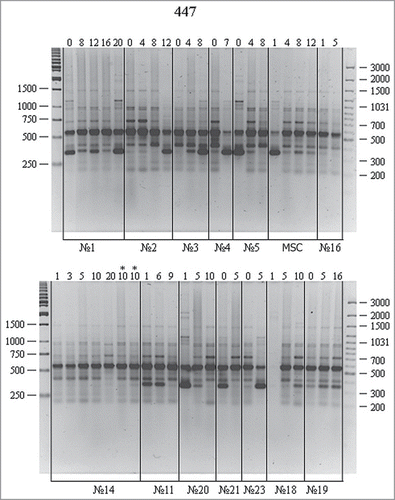
shows the amplification of the same DNA samples using the P447 primer. RAPD spectra display 6–8 fragments (including 2–4 major ones) in the electrophoretic zone of 250–1500 bp. At the background of a pool of major and minor bands with similar electrophoretic mobility, one can see significant RAPD variations both between cell cultures and passages of certain cultures. The amplified band of about 350 bp is the most illustrative. This fragment can be found as a major reproducible band in most cell cultures but is missing in culture nos. 16 and 14 and represented by a minor band in culture nos. 18 and 19. Reproducible differences are also observed for this band in different passages of the same cell culture. It could become more (culture nos. 2, 3, and 23) or less pronounced (MSCs and culture nos. 5, 11, 20, and 29) with passages.
Figure 3. RAPD analysis of DNA from cell cultures at different passages using the P447 primer. For designations, see .
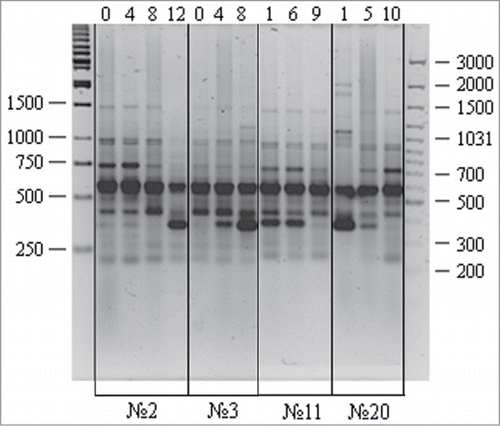
presents RAPD spectra for zero, intermediate, and final passages of several cell cultures. This applies to the bands of about 350 and 750 bp that demonstrate gradual changes at different culture passages. In the context of possible factors underlying the observed RAPD variations, one should realize that the studied cell cultures have different origin (mesenchymal, progenitor, adipose tissue, and skin fibroblasts), were obtained from different patients, and have heterogeneous cell composition. Accordingly, RAPD variations between zero passages of different cell cultures can be due to individual DNA variations of cell donors, while RAPD alterations in cell cultures with passages could result from structural DNA variations or changes in the proportion of cell fractions with structurally different DNA. Clearly, the analysis of the molecular structure of RAPD fragments can shed light on the nature of their possible variations with cell passages.
Figure 4. RAPD analysis of DNA isolated from different human cell cultures at different passages: variations observed for the P447 primer. A 100 bp Ladder+ and a 1 kb Ladder (Fermentas) were used as molecular weight markers. Culture nos. 2 and 3, adipose cells; nos. 11 and 20, skin fibroblasts; lanes 1–12, different passages.
Sequencing of RAPD fragments
Reproducible major amplification fragments obtained using the P447 primer in culture nos. 2, 3, 5 (adipose-derived stromal cells), and 11 (skin fibroblasts) were selected for sequencing (, framed).
Figure 5. RAPD analysis of DNA isolated from different human cell cultures at different passages: variations were observed for the P447 primer. A 100 bp Ladder+ and a 1 kb Ladder (Fermentas) were used as molecular weight markers. Culture nos. 2, 3, and 5, adipose-derived stromal cells; no. 11, skin fibroblasts; lanes 1–12, different passages. RAPD bands used in further experiments are marked by white frame.
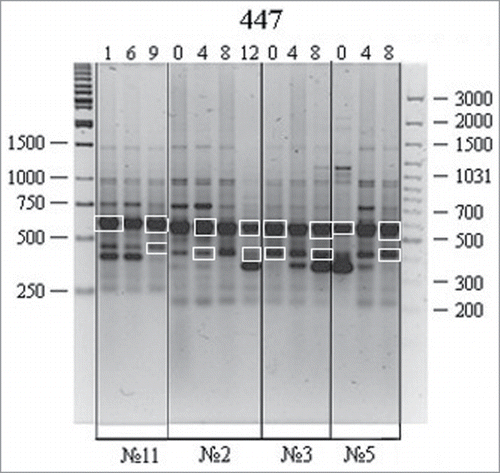
The amplification fragments were isolated from agarose gel, cloned, and sequenced as described in the Materials and Methods. For data authenticity, the whole procedure from DNA amplification and agarose gel electrophoresis to DNA cloning and sequencing was replicated twice. In case of data discrepancy, extra sequencing of certain fragments was performed. The obtained sequences corresponding to 3 different loci were analyzed using GenBank. The 548-, 388-, and 345-bp RAPD fragments were localized to chromosomes 8, 15, and 10, respectively (). Comparison of the sequences for different passages and cell cultures allowed us to identify randomly distributed single-nucleotide mismatches. An exception was found for the 548-bp fragment localized to chromosome 8 (8p23-p22, FAM167A region) with single-nucleotide variations (232 C→A and 307 C→T) at the same positions in several but not all cell cultures. Overall, no cell culture-specific variations have been identified.
Table 1. Chromosomal localization and single-nucleotide variations in RAPD fragments from human cell lines at different passages
Discussion
Therapeutic application of stem/progenitor cell culture poses significant requirements on the safety of cell material for humans. Abnormal changes at the levels of cell DNA, RNA, and proteins were demonstrated after long-term culture.Citation17 Cell culture assumes the isolation of stem cells, which impairs their natural environment and division control. In addition, cell culture selects for the cells that maintain active and long-term proliferation. High differentiation potential becomes disadvantageous for a cell in culture. Accordingly, stem cells in culture without the natural environment should have high proliferative activity and low differentiation potential. Such properties cannot but induce substantial alterations of the cells; furthermore, active uncontrolled proliferation can cause diseases (including cancer) in patients. Being aware of the abnormality of cell culture, researchers in the field studied abnormalities in cultured cells using various immunocytochemical and molecular methods. Chromosomal abnormalities and telomere shortening was demonstrated after long-term cell culture.Citation18 The biomarkers of cell senescence and accompanying abnormalities have also been studied.Citation19 In addition, studies of the proteomic profile of cultured cells also showed significant changes after the 5th passage.Citation20,21 Quantitative proteomic analysis involving 2D electrophoresis demonstrated substantial alterations in the proteomic profile in cells after 7 passages vs. those after 3 passages.Citation22 Of particular interest are the identified alterations in the profiles of cytoskeleton-associated proteins, vimentin isoforms, chaperone proteins, and apoptotic factors.Citation22 It goes without saying that such alterations cannot take place within a short period of one or 2 passages. Apparently, the changes at the level of DNA precede those at the level of proteins.
In this work, we used RAPD analysis to evaluate DNA stability/variation in human cell culture. This method is simple and fast but requires efficient primers and control for data reproducibility. In a previous study of somatic mosaicism in humans, we have found primers (P29, R45, and P447) that allowed informative RAPD spectra for human DNA samples.Citation16 The present work demonstrated that the P447 primer can be used for genomic identification and detection of genomic alterations in cells after long-term culture. Significant RAPD variations have been revealed between passages of certain cultures at the background of major and minor bands with similar electrophoretic mobility. For instance, reproducible distinctions in the content of the ∼350-bp amplification product were observed in different passages of the same cell culture. This band could become more or less pronounced with passages. Such RAPD variations can be due to genomic rearrangements, mutations, or epigenetic modifications covering the DNA regions complementary to the primers, as well as to DNA insertions and deletions. Further sequence analysis of the 3 RAPD fragments identified their chromosomal localization, while comparison of their sequences from different cell cultures and at different passages revealed numerous randomly distributed single-nucleotide variations. One of fragments 548 bp in length (8p23-p22 locus, FAM167A region) demonstrated variation hotspots (232 C→A and 307 C→T) in different cell cultures in addition to randomly distributed variations.
Experimental replicates exclude sequencing and PCR errors as a source of single-nucleotide variations. Rather they reflect de novo point mutations at these loci. GenBank mining also demonstrated 232 C→A and 307 C→T variation hotspots in the human genome. Other random variations that appeared in cells upon culturing are not limited to specific genomic sites or cell cultures.
The data obtained indicate early DNA modifications in cultured cells, which suggests that such modifications should be monitored. This study based on RAPD analysis demonstrates the genomic stability in cell cultures after no more than 5 passages. Human adipose-derived stromal cells showed high instability. Fibroblasts proved to be the most stable, and the stability varied between different fibroblast cultures. Thus, each cell culture deserves individual approach and genomic modifications should be monitored after long-term culture.
Materials and Methods
Biological material
Adipose tissue was obtained from abdominal surgery in non-oncological patients; human skin fibroblasts and primary cultures of human mesenchymal progenitor cells (MSCs) were kindly provided by Dr. Katerina A. Rubina (Lomonosov Moscow State University, Russia).
Fibroblast culture
Dermal fibroblasts were cultured in the fibroblast growth medium (DMEM, 10% FCS, 10 U/ml penicillin (Gibco), 10 U/ml streptomycin (Gibco), and 100 U/ml fungizone (Gibco)) in plastic flasks at 37°C in 5% CO2. After reaching confluence, cells were harvested by 0.25% trypsin and 0.02% EDTA (Gibco). Detached cell suspension was plated at the ratio of 1:3.
Isolation of stromal cells from adipose tissue
Human adipose-derived stromal cells (ASCs) used in this work were obtained from non-oncological patients with informed consent approved by the Ethics Committee of Moscow State University (Permit Number: 45.01/06.11.2013). It was carried out in strict accordance with the ethical rules of animal experimentation in accord with the European Community Council Directives (86/609/EEC and 2010/63/EU). Cells were isolated according to BruderCitation23 with modifications. The used method provided for the yield of 5–7 × 105 cells per 30 ml adipose tissue cells obtained after the liposuction. After reaching 80% confluence, cells were harvested with Versene, 0.25% trypsin, and 0.02% EDTA, and cultured. Cultured cells demonstrated no decrease in the division rate. They had no explicit contact inhibition and continued to divide after confluence although at a lower rate. When replating, one portion was used for DNA isolation and another one was cultured, and so on until the final passage.
DNA isolation from tissues
Tissue samples stored at −70°C were ground to powder in liquid nitrogen and transferred to 2–4 ml of buffer 2 containing 75 mM NaCl, 25 mM EDTA, and 25 mM Трис-HCl (pH 8.0). The cells were lysed with 1% SDS and 50–100 μg/ml proteinase K (Sigma) at 37°C overnight. The lysate was supplemented with 4 M sodium acetate to 0.2 M and gently agitated. After phenol extraction and ethanol precipitation, the DNA was dissolved in TE and its concentration was measured by spectrophotometry at 260 nm. DNA solution was stored at +4°C.
DNA isolation from cell culture
Cells were harvested by centrifugation at 1000 rpm for 10 min at +4°C. The pellet was broken by tapping the tube bottom, supplemented with ∼12 ml of Earle's medium, resuspended with a serological pipette, and transferred to a 50 ml tube. The cells were lysed with 1% SDS and 50–100 μg/ml proteinase K (Sigma) at 37°C overnight. The lysate was supplemented with 4 M sodium acetate to 0.2 M and gently agitated. After phenol extraction and isopropanol precipitation, the DNA was dissolved in TE and its concentration was measured by spectrophotometry at 260 nm. DNA solution was stored at +4°C.
Plasmid DNA isolation
Plasmid DNA was isolated using a GeneJet Plasmid Miniprep Kit (Fermentas, USA) according to the manufacturer's instructions.
Polymerase chain reaction
PCR was carried out in 20 μl reactions containing 100 ng of genomic DNA and 1 mM of each primer using a GenePak PCR Core kit (Isogene Lab, Russia) according to the manufacturer's instructions. Amplification was performed in a 4-channel DNA amplifier Tertsik TP4-PCR-01 (DNA-Technology, Russia) using the following program: 94°С for 3 min; 40 cycles of 93°С for 10 s, 38°С (P29 and R45) or 40°C (P447) for 40 s, and 72°С for 3 min; and final 72°С for 5 min. The primers used were P29 (5′-CCGGCCTTAC-3′), R45 (5′-GCCGTCCGAG-3′), and P447 (5′-AACGGTCACG-3′). PCR products were analyzed by electrophoresis in 3% agarose gel using the video documentation system BioDocAnalyze (Biometra, Germany).
DNA elution from agarose gel
DNA fragments were isolated from agarose gel using a GFX PCR DNA and Gel Band Purification Kit (Amersham) according to the manufacturer's instructions.
DNA cloning
DNA cloning was performed using a pGEM-T Easy Vector System I (Promega, United States) according to the manufacturer's instructions.
Sequencing of PCR products
DNA sequences were determined by the dideoxy chain termination method using an ABI PRISM BigDye Terminator v.3.1 Cycle Sequencing Kit (GE Healthcare, United States) and a 3730 DNA Analyzer (Applied Biosystems, USA).
Computer analysis of nucleotide sequences
The obtained sequences were analyzed using the DNASTAR programs (DNASTAR, United States). Sequence alignments were generated with the Clustal W algorithm implemented in the MEGA 6.0.5 software (http://megasoftware.net).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Our thanks are due to Dr. Dmitri Yu. Panteleev for his help in culturing adipose-derived stromal cells. We also thank Dr. Katerina A. Rubina for providing fibroblast cell culture.
Funding
This work was partially supported by the grant Russian Foundation for Basic Research (project no. 13-04-40200H, 13-00-40200K), the Program of the Russian Academy of Sciences for Molecular and Cell Biology, and the President RF Program of Leading Scientific Schools (ss-2501.2014.4).
References
- Tarte K, Gaillard J, Lataillade JJ, Fouillard L, Becker M, Mossafa H, Tchirkov A, Rouard H, Henry C, Splingard M, et al. Clinical-grade production of human mesenchymal stromal cells: occurrence of aneuploidy without transformation. Blood 2010; 115(8):1549-53; PMID:20032501; http://dx.doi.org/10.1182/blood-2009-05-219907
- Sensebé L, Bourin P, Tarte K. Good manufacturing practices production of mesenchymal stemstromal cells. Hum Gene Ther 2011; 22(1):19-26; http://dx.doi.org/10.1089/hum.2010.197
- Ueyama H, Horibe T, Hinotsu S, Tanaka T, Inoue T, Urushihara H, Kitagawa A, Kawakami K. Chromosomal variability of human mesenchymal stem cells cultured under hypoxic conditions. J Cell Mol Med 2012; 16(1):72-82; PMID:21418515; http://dx.doi.org/10.1111/j.1582-4934.2011.01303.x
- Mikkola M, Olsson C, Palgi J, Ustinov J, Palomaki T, Horelli-Kuitunen N, Knuutila S, Lundin K, Otonkoski T, Tuuri T. Distinct differentiation characteristics of individual human embryonic stem cell lines. BMC Dev Biol 2006;8:6-40; PMID:16895598
- Pradel W, Mai R, Gedrange T, Lauer GJ. Cell passage and composition of culture medium effects proliferation and differentiation of human osteoblast-like cells from facial bone. Physiol Pharmacol 2008; 59(5):47-58; PMID:19075324
- Nguyen TD, Widera D, Greiner J, Müller J, Martin I, Slotta C, Hauser S, Kaltschmidt C, Kaltschmidt B. Prolonged cultivation of hippocampal neural precursor cells shifts their differentiation potential and selects for aneuploid cells. Biol Chem 2013; 394(12):1623-36; PMID:24084358; http://dx.doi.org/10.1515/hsz-2013-0191
- Reinoso E, Bettera S, Frigerio C, DiRenzo M, Calzolari A, Bogni C. RAPD-PCR analysis of Staphylococcus aureus strains isolated from bovine and human hosts. Microbiol Res 2004; 159(3):245-55; PMID:15462524; http://dx.doi.org/10.1016/j.micres.2004.04.002
- Devaiah KM, Venkatasubramanian P. Genetic characterization and authentication of Embelia ribes using RAPD-PCR and SCAR marker. Planta Med 2008; 74(2):194-6; PMID:18210350; http://dx.doi.org/10.1055/s-2008-1034279
- Munoz MA, Welcome FL, Schukken YH, Zadoks RN. Molecular epidemiology of two Klebsiella pneumoniae mastitis outbreaks on a dairy farm in New York State. J Clin Microbiol 2007; 45(12):3964-71; PMID:17928424; http://dx.doi.org/10.1128/JCM.00795-07
- Lee YC, Yang VC, Wang TS. Use of RAPD to detect sodium arsenite-induced DNA damage in human lymphoblastoid cells. Toxicology 2007; 239(1-2):108-15; PMID:17707572; http://dx.doi.org/10.1016/j.tox.2007.06.101
- Dil-Afroze, Misra A, Sulaiman IM, Sinha S, Sarkar C, Mahapatra AK, Hasnain SE. Genetic alterations in brain tumors identified by RAPD analysis. Gene 1998; 206(1):45-8; PMID:9461413; http://dx.doi.org/10.1016/S0378-1119(97)00579-9
- Ong TM, Song B, Qian HW, Wu ZL, Whong WZ. Detection of genomic instability in lung cancer tissues by random amplified polymorphic DNA analysis. Carcinogenesis 1998; 19(1):233-5; PMID:9472718; http://dx.doi.org/10.1093/carcin/19.1.233
- Calvisi DF, Factor VM, Ladu S, Conner EA, Thorgeirsson SS. Disruption of beta-catenin pathway or genomic instability define two distinct categories of liver cancer in transgenic mice. Gastroenterology 2004; 126(5):1374-86; PMID:15131798; http://dx.doi.org/10.1053/j.gastro.2004.02.014
- Xian ZH, Cong WM, Zhang SH, Wu MC. Genetic alterations of hepatocellular carcinoma by random amplified polymorphic DNA analysis and cloning sequencing of tumor differential DNA fragment. World J Gastroenterol 2005; 11(26):4102-7; PMID:15996039
- Luceri C, De Filippo C, Caderni G, Gambacciani L, Salvadori M, Giannini A, Dolara P. Detection of somatic DNA alterations in azoxymethane-induced F344 rat colon tumors by random amplified polymorphic DNA analysis. Carcinogenesis 2000; 21(9):1753-6; PMID:10964108; http://dx.doi.org/10.1093/carcin/21.9.1753
- Butovskaia PR, Martirosian IA, Baranov VS, Egorova AA, Kiselav AV, Pavlova GV, Korochkin LI. Detection of somatic mosaicism in humans using polymerase chain reaction with random primers. Genetika 2007; 43(12):1694-99; PMID:18592698
- Oliveira PH, Boura JS, Abecasis MM, Gimble JM, da Silva CL, Cabral JM. Impact of hypoxia and long-term cultivation on the genomic stability and mitochondrial performance of ex vivo expanded human stemstromal cells. Stem Cell Res 2012; 9(3):225-36; PMID:22903042; http://dx.doi.org/10.1016/j.scr.2012.07.001
- Kim J, Kang JW, Park JH, Choi Y, Choi KS, Park KD, Baek DH, Seong SK, Min HK, Kim HS. Biological characterization of long-term cultured human mesenchymal stem cells. Arch Pharm Res 2009; 32:117-26; PMID:19183884; http://dx.doi.org/10.1007/s12272-009-1125-1
- Kim JS, Kim EJ, Kim HJ, Yang JY, Hwang GS, Kim CW. Proteomic and metabolomic analysis of H2O2-induced premature senescent human mesenchymal stem cells. Exp Gerontol 2011; 46:500-10; PMID:21382465; http://dx.doi.org/10.1016/j.exger.2011.02.012
- Feldmann RE Jr, Bieback K, Maurer MH, Kalenka A, Burgers HF, Gross B, Hunzinger C, Klüter H, Kuschinsky W, Eichler H. Stem cell proteomes: a profile of human mesenchymal stem cells derived from umbilical cord blood. Electrophoresis 2005; 26:2749-58; PMID:15971194; http://dx.doi.org/10.1002/elps.200410406
- Foster LJ, Zeemann PA, Li C, Mann M, Jensen ON, Kassem M. Differential expression profiling of membrane proteins by quantitative proteomics in a human mesenchymal stem cell line undergoing osteoblast differentiation. Stem Cells 2005; 23:1367-77; PMID:16210410; http://dx.doi.org/10.1634/stemcells.2004-0372
- Madeira A, da Silva CL, dos Santos F, Camafeita E, Cabral JM, Sa´-Correia I. Human mesenchymal stem cell expression program upon extended ex-vivo cultivation, as revealed by 2-DEBased quantitative proteomics. PLoS One 2012; 7(8):e43523; PMID:22916271; http://dx.doi.org/ 10.1371journal.pone.0043523
- Bruder CE, Piotrowski A, Gijsbers AA, Andersson R, Erickson S, Diaz de Ståhl T, Menzel U, Sandgren J, von Tell D, Poplawski A, et al. Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am J Hum Genet 2008; 82:763-71; PMID:18304490; http://dx.doi.org/10.1016/j.ajhg.2007.12.011