
Abstract
Cellular supply of deoxynucleoside triphosphates (dNTPs) is crucial for DNA replication and repair. In this study, we investigated the role of CMP/UMP kinase (CMPK), an enzyme catalyzes CDP formation, in DNA repair. Knockdown of CMPK delays DNA repair during recovery from UV damage in serum-deprived cells but not in the cells without serum deprivation. Exogenous supply of cytidine or deoxycytidine facilitates DNA repair dependent on CMPK in serum-deprived cells, suggesting that the synthesis of dCDP or CDP determines the rate of repair. However, CMPK knockdown does not affect the steady state level of dCTP in serum-deprived cells. We then found the localization of CMPK at DNA damage sites and its complex formation with Tip60 and ribonucleotide reductase. Our analysis demonstrated that the N-terminal 32-amino-acid of CMPK is required for its recruitment to DNA damage sites in a Tip60-dependent manner. Re-expression of wild-type but not N-terminus deleted CMPK restores the efficiency of DNA repair in CMPK knockdown cells. We proposed that site-specific dCDP formation via CMPK provides a means to facilitate DNA repair in serum-deprived cells.
Abbreviations
| dNTP | = | deoxynucleoside triphosphates; CMPK, CMP/UMP kinase; RNR, ribonucleotide reductase; 6-4 pp, (6-4) photoproduct; R1C, C-terminal fragment of R1 subunit of RNR |
Introduction
Cellular supply of deoxyribonucleoside triphosphates (dNTPs) is critical for DNA synthesis during replication and repair. The synthesis of dNTPs is cell-cycle regulated, reaching the highest in the S phase and the lowest in quiescent state.Citation1,2 Since DNA damage can occur in all phase of cell cycle, it is interesting to know the relation of dNTPs synthesis with DNA repair in quiescent and proliferating cells. We have previously shown that salvage synthesis of dTTP in mitochondria plays an important role in determining the rate of DNA repair in quiescent fibroblasts.Citation3 Unlike dTTP, the synthesis of dCTP, dATP and dGTP in quiescent cells is highly dependent on the activity of ribonucleotide reductase (RNR).Citation4 Mammalian RNR is a tetrameric complex composed of 2 large catalytic (R1) subunits and 2 small (R2 or p53R2) subunits. The level of R2 expression is cell-cycle regulated, which is responsible for high dNTP pools in the S phase.Citation5-7 RNR catalyzes the formation of these dNDPs through the reduction of the corresponding NDPs. These RNR substrates, NDPs—ADP, GDP, CDP and UDP, are generated by AMP kinase, GMP kinase and UMP/CMP kinase (CMPK) from corresponding NMPs, respectively.Citation8-10 Human CMPK has been identified (EC 2.7.4.14) and biochemically characterized. CMP and UMP are the preferred substrates for purified CMPK, and it has lower affinity to dCMP and dUMP.Citation11 In this study, the functional contribution of CMPK to dCTP pool and the rate of DNA repair were compared in MCF-7 cells in serum-containing and serum-deprived conditions. Our data showed the differential contribution of CMPK to the rate of DNA repair in these conditions, which are related to the steady-state level of dCTP. Intriguingly, in serum-deprived cells that have low level of dCTP, CMPK knockdown delays DNA repair without affecting the pool of dCTP.
It has been reported that the R1 subunit of RNR interacts with Tip60, which is a histone acetyltransferase. In response to DNA damage, Tip60 is activated by binding with lysine 9 trimethylation of histone 3 to activate ATM and acetylate histone to remodel chromatin structure for repair.Citation12-14 Through the interaction with Tip60, RNR is recruited to DNA damage sites for local supply of dNTPs. This process is essential for DNA repair in G1 phase of mammalian cells, which contain low steady-state level of dNTP pools.Citation15,16 Our laboratory has previously found that thymidylate kinase, which catalyzes the reaction of dTDP formation, is also enriched at DNA damage sites to coordinate with RNR for repair.Citation17 In addition, the enzymes of de novo thymidylate synthesis pathway form a scaffolded multienzyme complex at replication sites in the nucleus.Citation18 These studies demonstrated that site-specific synthesis of dNTPs provides an efficient way to ensure sufficient building blocks for DNA synthesis during repair and replication. In this study, we present evidence that CMPK forms a complex with Tip60/RNR, and is recruited to DNA damage sites. CMPK mutant defective in this complex formation and localization at DNA damage sites has no function in facilitating DNA repair in serum-deprived cells. According to our data, we proposed that compartmentalized synthesis of dCDP at DNA damage sites affects the rate of DNA repair in cells to complement low dCTP supply from cytosol in the serum-deprived condition.
Results
Differential contribution of CMPK in repairing UV-induced DNA lesions in serum-containing and -deprived conditions
In order to investigate the functional contribution of CMPK to DNA repair and its influence on cellular level of dCTP, CMPK was depleted by lentiviral shRNA infection in MCF-7 cells. It has been reported that MCF-7 is responsive to serum deprivation to give more than 70% population accumulated in the G1 phase.Citation19 We compared the effects of CMPK knockdown on dNTP pools in serum-containing and serum-deprived conditions. CMPK knockdown decreased dCTP pool by 44% while having no effect on other 3 dNTP levels in serum-containing condition. The levels of all 4 dNTP pools were significantly decreased after serum deprivation, and CMPK knockdown did not additionally reduced the dCTP level (). Although dNTP pools were reduced by serum deprivation, the expression level of RNR was not reduced (). Cell cytometric analysis indicated a reciprocal alteration in cells distributed in G2/M and G0/G1 fractions after serum deprivation, and CMPK knockdown did not alter the cell cycle profile (). These cells were exposed to UV irradiation. It has been shown that whole-cell UV irradiation generates gaps during nucleotide excision repair (NER)-mediated lesion removal, which are then turned to γH2AX foci.Citation3,20 Under the serum-containing condition, strong γH2AX immunofluorescence staining signals appeared at 4 h and disappeared at 8 h in these cells after recovery from UV irradiation, and CMPK knockdown had no effect on the intensity and the temporal change in γH2AX signal (). However, under the serum-deprived condition, after recovery from UV irradiation for 4 h, CMPK-depleted cells exhibited higher intensity of γH2AX signal than that of control cells. The lesion signals became diminished at 8 h recovery in control cells, while in CMPK-depleted cells, γH2AX staining remained sustained (). It was until 16 h that γH2AX staining largely decreased in CMPK knockdown cells (). The viability assay showed no difference in the survival between control and CMPK knockdown cells after UV irradiation (Fig. S1). Thus, CMPK knockdown specifically affects the rate of DNA repair, not the survival, in the serum-deprived cells.
Figure 1. The functional contribution of CMPK to dCTP pool and DNA repair in serum-containing and -deprived conditions. (A–C) MCF-7 cells were infected with Luciferase (Luc) or CMPK shRNA lentivirus. These cells were incubated with medium containing 10% serum (serum-containing) or serum-free (serum-deprived) for 72 h and harvested for dNTP pool determination (n = 3) (A), protein gel blot with indicated antibodies (B), and propidium iodide staining to analyze cell cycle by flow cytometry (n =3) (C). Below the Western blot, the relative level of R2 was shown, which was quantified in 3 independent experiments by GELPRO32 and indicated as mean ± s.d. (D and E) Control (shLuc) or CMPK knockdown (shCMPK) MCF-7 cells were whole-cell irradiated by 15 J/M2 UVC and fixed at indicated time points for γH2AX immunofluorescence staining in serum-containing (D) or serum-deprived condition (E). Fluorescence intensity was expressed relative to that in shLuc cells recovered at 4 h. 50 cells were analyzed for each experiment (n = 3). (F and G) Control and CMPK knockdown MCF-7 cells were serum-deprived for 72 h and irradiated by 120 J/M2 UVC through micro-pore membrane and fixed at indicated time points for 6-4 photoproducts (6-4 pp) immunofluorescence staining (F) or PCNA immunofluorescence staining (G). Fluorescence intensity was expressed relative to that in shLuc cells recovered at 0.5 h. 50 cells were analyzed for each experiment (n = 3). Scale bar, 10 μm. Error bars represent s.d., * P < 0.05, ** P < 0.01, *** P < 0.001 based on Student's t-test.
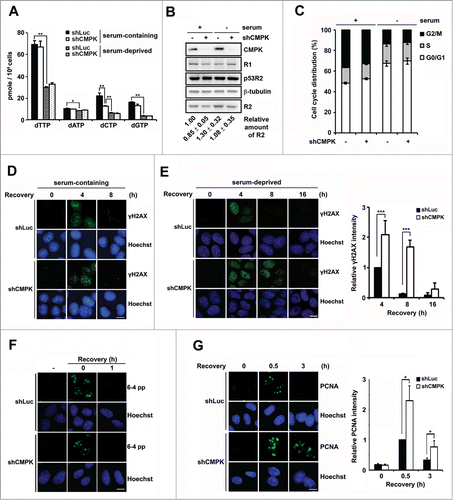
UV irradiation causes DNA lesions resulting from cyclobutane pyrimidine dimer and (6-4) photoproduct (6-4 pp) formation.Citation21 In mammalian cells, these lesions in genomic DNA are repaired by nucleotide excision repair (NER) pathway. After lesion recognition, the excision step in NER process causes a single-stranded DNA gap, which requires 24–32 deoxynucleotides incorporation in each gap to complete the repair.Citation22 To know whether CMPK knockdown in serum-deprived cell affects the step of removal of gap-filling, we performed UV micro-irradiation by applying UV irradiation through a 3 μm-pore filter paper to analyze the lesion removal and gap-filling steps of NER in serum-deprived MCF-7 cells. After micro-irradiation, the signals of 6-4 pp immediately appeared, and then rapidly diminished within 1 h. No difference in the removal of 6-4 pp was observed in control and CMPK knockdown cells (). The gap-filling step in UV micro-irradiated lesion was measured by PCNA immunofluorescence staining.Citation3 We found that the intensity of PCNA foci signal was stronger in CMPK-depleted cells at 0.5 h post-irradiation. After 3 h, the signals of PCNA foci disappeared in control cells, while in CMPK-depleted cells the signals were still detectable (). This indicates that the completion of gap-filling step is slowed down by CMPK depletion in serum-deprived cells. Here, it should be emphasized that the amounts of genome lesions generated by UV micro-irradiation and whole-cell irradiation are different. The signals of γH2AX foci were generated from whole-cell irradiation, because more lesions all over the nuclear genome required longer time and more dNTPs for repair.Citation3,20,23,24
Cytidine and deoxycytidine facilitate DNA repair dependent on CMPK in serum-deprived cells
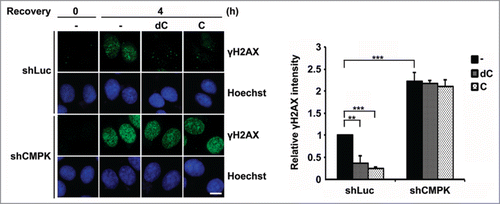
Although it has been shown that the preferred substrate of purified CMPK is CMP not dCMP, the CMPK enzyme activity in different cell lysates was similar in the utilization of UMP, CMP and dCMP as the substrates.Citation11,25 We further analyzed the contribution of direct CDP or dCDP formation from CMPK to DNA repair in serum-deprived cells. To this end, we added cytidine and deoxycytidine in serum-free medium, which presumably increased the cellular level of CMP and dCMP, respectively, via the salvage kinases.Citation26,27 We then compared DNA repair in control and CMPK knockdown cells under serum-deprived condition. After recovery for 4 h from UV exposure, the strong γH2AX signals in control cells were very clear. Aforementioned data in showed that these lesion signals did not disappear until recovery for 8 h. Here, exogenous addition of cytidine or deoxycytidine in medium abolished the lesion signals at 4 h recovery, suggesting each input significantly increases the rate of DNA repair (). However, these effects were not seen in CMPK knockdown cells (). Thus, CMPK appears to be critical in mediating CDP or dCDP for DNA repair in serum-deprived MCF-7 cells.
Figure 2. The effects of exogenous supplement of cytidine and deoxycytidine on DNA repair via CMPK in serum-deprived cells. Control (shLuc) or CMPK knockdown (shCMPK) MCF-7 cells were serum-deprived for 3 days and preincubated with 5 μM deoxycytosine (dC) or cytosine (C) for 8 h before UV irradiation. After recovery with or without 5 μM dC or C for 4 h, cells were fixed for γH2AX immunofluorescence staining. Scale bar, 10 μm. Fluorescence intensity was expressed relative to that in shLuc cells without dC and C. 50 cells were analyzed for each experiment (n = 3), error bars represent s.d., **, P < 0.01, ***, P < 0.001 based on Student's t-test.
The complex of CMPK/RNR/Tip60 and the recruitment to the sites of DNA damage
CMPK has a functional role in DNA repair in serum-deprived cells that contain low level of dCTP level. As CMPK knockdown did not further decrease the pool in these cells, we speculated the involvement of the local function of CMPK at DNA damage sites in repair. To this end, we transfected cells with expression vector of flag-CMPK and performed laser micro-irradiation to generate local DNA lesions. We used HeLa cells for laser micro-irradiation because of their large nuclear size. The immunofluorescence staining showed the presence of flag-CMPK co-localized with γH2AX at the irradiated lesion lines. Interestingly, we found the large subunit R1 of RNR also co-localized with flag-CMPK at DNA damage sites (). Given that RNR is recruited to DNA damage sites through interaction with Tip60, we asked whether the recruitment of flag-CMPK is also Tip60-dependent. To this end, we performed lentiviral Tip60 shRNA infection to decrease the level of Tip60 expression in HeLa cells. The knockdown effect was assured by RT-qPCR because the poor sensitivity of Tip60 antibody in western blotting of endogenous Tip60 (). The recruitment of flag-CMPK to the laser-irradiated line was reduced by Tip60 knockdown, so as endogenous R1 ( and ). Since Tip60 knockdown did not change the expression level of flag-CMPK (), these results indicate the essential and common role of Tip60 in CMPK and RNR localization at DNA damage sites.
Figure 3. (See previous page) Tip60/RNR-dependent CMPK recruitment to DNA damage sites. (A) HeLa cells were transfected with flag-CMPK followed by laser micro-irradiation. After recovery for 5 min, cells were fixed for immunofluorescence staining with indicated antibodies. (B–D) Flag-CMPK-transfected HeLa cells were infected with LacZ or Tip60 shRNA lentivirus. After 48 h infection, cells were harvested for RT-qPCR for detecting Tip60 mRNA expression which was shown as percentage relative to LacZ shRNA control (n = 3, error bar represent s.d.) and the expression of flag-CMPK was detected by western blot (B). These cells were plated on glass-bottomed dishes for laser micro-irradiation. After recovery for 5 min, cells were fixed for immunofluorescence staining with γH2AX and R1 (C) or M2 (D) antibodies. Scale bar, 10 μm. Percentage of flag-CMPK colocalized with γH2AX was shown as mean ± s.d. (n = 3). More than 30 cells with localized γH2AX were analyzed in each experiment, *, P < 0.05 based on Student's t-test. (E and F) HEK 293T cells transfected with HA-Tip60 and flag-CMPK were prepared for immunoprecipitation by HA antibody-conjugated beads (E) or M2 beads (F) followed by protein gel blot with indicated antibodies. (G) HeLa cells were transfected with flag-CMPK and mCherry vector (−) or mCherry-NLS-R1C (+) in a ratio of 3: 1. The mCherry positive cells were performed laser micro-irradiation. After recovery for 5 min, cells were fixed for immunofluorescence staining with M2 and γH2AX antibodies. Scale bar, 10 μm. The expression of mCherry-NLS-R1C and flag-CMPK were detected by western blot. Percentage of flag-CMPK colocalized with γH2AX was shown as mean ± s.d. (n = 3). More than 30 cells with localized γH2AX were analyzed in each experiment, **, P < 0.01 based on Student's t-test.
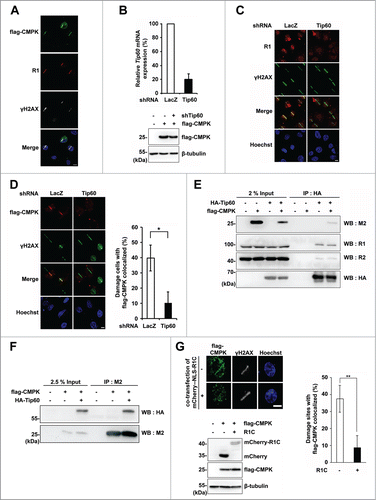
Next, we accessed the possibility of complex formation between CMPK and Tip60/RNR. We overexpressed HA-Tip60 and flag-CMPK in HEK 293T cells that have high transfection efficiency. The results from co-immunoprecipitation experiment by HA antibody showed that overexpression of HA-Tip60 formed a complex with endogenous RNR subunits and flag-CMPK (). Reciprocal co-immunoprecipitation by flag antibody, M2, also gave a similar result (), suggesting that flag-CMPK is capable of interacting with Tip60/RNR complex. It is already known that RNR recruited to DNA damage sites is dependent on the specific interaction of C-terminal 90 amino acids of R1 subunit with Tip60.Citation12 To know whether RNR is responsible for the recruitment of flag-CMPK to DNA damage sites, cells were transfected with expression vector of C-terminal fragment of R1 (R1C) to disrupt the specific interaction of RNR with Tip60, thereby intervening the recruitment of endogenous RNR at sites of DNA damage. Using laser micro-irradiation, we found that expression of mCherry-R1C reduced the localization of flag-CMPK to the DNA damage site (). Knockdown of R1 also gave similar results (Fig. S2). These results indicate that CMPK recruitment to the DNA damage site is dependent on RNR interaction with Tip60.
The N-terminal region of CMPK is essential for the complex formation and recruitment to DNA damage sites
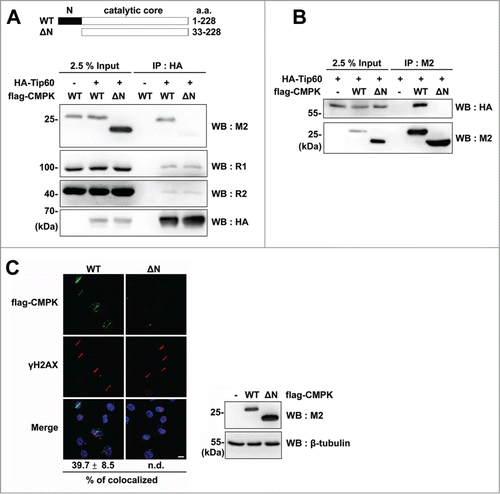
Given the interaction of Tip60 with CMPK, we further analyzed the region of CMPK required for complex formation. We expressed full-length of CMPK and its N-terminal 32 amino acids-deleted form which retains the catalytic domain in HEK 293T cells ().Citation11 The co-immunoprecipitation experiment showed that the expression of N-terminal 32 amino acids-deleted form (ΔN) was unable to form a complex with HA-Tip60 but had no effect on HA-Tip60/RNR complex formation (). Reciprocal co-immunoprecipitation by M2 beads also showed no complex formation of flag-CMPK mutant (ΔN) with HA-Tip60 (). These results reveal the essential role of N-terminal region of CMPK in the complex formation between Tip60/RNR and CMPK. We further tested whether N-terminal region is required for CMPK localization at DNA damage sites by laser micro-irradiation experiment. In accordance, flag-CMPK mutant (ΔN) was not localized at the DNA damage sites (). Altogether, these data suggest that the N-terminal region of CMPK mediates the complex formation with Tip60/RNR, by which CMPK is co-recruited to DNA damage sites.
Figure 4. N-terminal region of CMPK is necessary for the complex formation with Tip60/RNR and recruitment to DNA damage sites. (A and B) HEK 293T cells transfected with HA-Tip60 and flag-CMPK constructs were prepared for immunoprecipitation by HA antibody-conjugated beads (A) or M2 beads (B) followed by protein gel blot with indicated antibodies. WT: wild-type. ΔN: N-terminal 32 amino acids-deleted. (C) HeLa cells were transfected with wild-type or mutant form (ΔN) of flag-CMPK. These cells were plated on glass-bottomed dishes for laser micro-irradiation. After recovery for 5 min, cells were fixed for immunofluorescence staining with M2 and γH2AX antibodies. Scale bar, 10 μm. Percentage of flag-CMPK colocalized with γH2AX was shown as mean ± s.d. (n = 3). More than 30 cells with localized γH2AX were analyzed in each experiment, n.d.: non-detected. The expression of different forms of flag-CMPK was detected by western blot.
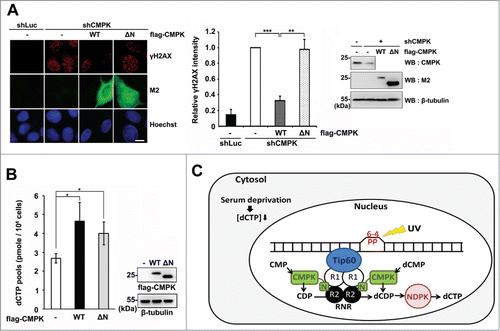
The N-terminal region of CMPK is required for the involvement of CMPK in DNA repair in serum-deprived cells
To know whether the complex formation of Tip60/RNR/CMPK plays a role in DNA repair, we reintroduced the expression constructs of wild-type and ΔN CMPK which are shRNA-resistant in CMPK knockdown cells. After serum deprivation followed by recovery from UV irradiation for 8 h, the results of γH2AX immunofluorescence staining showed that re-expression of wild-type, but not mutant form (ΔN) of flag-CMPK rescued DNA repair (). Overexpression of either wild-type or mutant form (ΔN) of flag-CMPK was able to expand dCTP pool in HEK 293T cells (). The analysis of CMP, CDP and CTP pools also showed that overexpression of either wild-type or mutant form (ΔN) of CMPK increased CTP pool (Table S1). These results indicate that the ΔN mutant is functionally intact in promoting the flux of dCTP/CTP production. Since the total dCTP pool was not affected by CMPK knockdown in serum-deprived cells, we proposed that the N-terminal region confers CMPK a function for Tip60/RNR/CMPK complex formation, by which DNA damage site-specific synthesis of dCDP has a functional contribution to the efficiency of DNA repair ().
Figure 5. The N-terminal region of CMPK has a functional contribution to DNA repair in serum-deprived MCF-7 cells. (A) Control (shLuc) or CMPK knockdown (shCMPK) MCF-7 cells were transfected with empty vector (-), wild-type (WT) or mutant form (ΔN) of shRNA-resistant flag-CMPK followed by serum deprivation and whole-cell UV irradiation (15 J/M2). After recovery for 8 h, cells were fixed for γH2AX immunofluorescence staining. Fluorescence intensity was expressed relative to that in shCMPK cells. Scale bar, 10 μm. 50 cells were analyzed for each experiment (n = 3). The expression of endogenous CMPK and flag-CMPK constructs were detected by protein gel blot. (B) HEK 293T cells were transfected with wild-type (WT) or mutant form (ΔN) of flag-CMPK. Then cells were harvested to determine cellular dCTP pools (n = 3). Error bars represent s.d., * P < 0.05, ** P < 0.01, *** P < 0.001 based on Student's t-test. (C) A proposed model of the cooperation of CMPK with Tip60/RNR at DNA damage sites for supply of dCTP, acting as a rate-limiting event for DNA repair in serum-deprived condition, which contains low cellular dCTP supply from cytosol. NDPK: nucleoside diphosphate kinase.
Discussion
In this report, UV irradiation was used to evaluate the functional role of CMPK in DNA repair. We showed that knockdown of CMPK significantly delays DNA repair in serum-deprived MCF-7 cells that contain low level of dCTP pool. In mammalian cells, UV-induced lesions are repaired by nucleotide excision repair, which process involves excision of UV-induced photoproducts followed by gap-filling of 24–32 newly synthesized deoxynucleotides incorporation by DNA polymerization. By detecting (6-4) photoproducts and PCNA foci in the nuclear genome upon UV micro-irradiation, we found that CMPK depletion not only intensified but also prolonged PCNA foci during recovery without affecting the removal of (6-4) photoproducts in serum-deprived cells. Since PCNA foci represent the gap-filling step of NER, this result suggests that CMPK has a functional contribution to the completion of gap-filling. Consistently, CMPK knockdown also caused the sustained γH2AX staining signal during recovery from whole-cell UV irradiation. Exogenous supply of either cytidine or deoxycytidine in serum-free medium was able to facilitate DNA repair. More importantly, CMPK knockdown abolished their effects. Thus, dCDP and CDP formation via CMPK can act as a rate-limiting factor in DNA repair in serum-deprived cells. A previous study has shown that pol κ, a translesion synthesis (TLS) polymerase with low replicative fidelity, is involved in gap-filling step of NER during the condition of low dNTP pools.Citation28 A recent study has identified a new TLS polymerase, PrimPol, which function in lesion bypass during replication is dNTP dependent.Citation29 In the future, it would be interesting to understand how the DNA polymerase is selected to adapt low dNTP pools for the gap-filling step in the NER process in quiescent cells.
Cytidine can be converted to CMP by uridine-cytidine kinase (UCK), while deoxycytidine is converted to dCMP by deoxycytidine kinase.Citation26,27 Presumably, CMPK then generates dCDP and CDP directly from dCMP and CMP, respectively. CDP would require additional step of reduction by RNR to form dCDP. In this study, we found that CMPK forms a complex with RNR and Tip60, which is co-localized at DNA damage sites. It is known that Tip60 is a histone acetyltransferase that regulates the DNA damage response by acetylating histone and remodeling chromatin at sites of DNA double-stranded beaks (DSBs) for repair.Citation12 Recent studies have shown that accumulation of un-repaired gaps after UV damage would be converted to double-stranded breaks with ATM activation signal in quiescent cells.Citation3,20 The interaction of R1 subunit of RNR with Tip60 allows RNR and CMPK to be recruited to DNA damage sites as a part of DNA damage response. Therefore, the functional coupling of CMPK and RNR leads to the conversion of CMP to dCDP at DNA damage sites, thus facilitating the supply of dCTP for repair. We defined the N-terminus of CMPK required for the complex formation. In support of our hypothesis, this mutant is defective in restoring DNA repair in CMPK knockdown cells under the serum-deprived condition. Accordingly, we proposed that site-specific dCDP formation either directly from CMPK or from CMPK/RNR offers an advantage for DNA repair (). Under the condition of high dCTP level, this pathway might be dispensable. In the serum-deprived condition, the presence of this mechanism complements the limiting supply of dCTP from the cytosolic pool by increasing the local synthesis for DNA repair.
It is noted that the amounts of R2, p53R2, R1 and CMPK proteins were not reduced in MCF-7 cells after serum deprivation. So, why dCTP level is decreased? We speculate that CDP synthesis might be decreased in serum-deprived cells, in turn reducing output of dCTP synthesis from RNR. It has been reported that the multienzyme, CAD (carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase) which is essential for CMP synthesis, is activated by serum stimulation.Citation30,31 Therefore, the production of dCTP from CMP through the subsequent action of CMPK, RNR and NDPK is serum-sensitive and responsive to CMPK depletion. In serum-deprived cells, however, depletion of CMPK protein did not reduce the dCTP pool. This indicates that the limiting synthesis of CMP in these cells lead to the net synthesis of dCTP through cytosolic CMPK so little. In addition, UMP/CMP kinase 2 (CMPK2) in mitochondria might provide an alternative contribution to dCTP in serum-deprived cells.Citation32 The synthesis of dCDP via CMPK recruitment to DNA damage sites is converted to dCTP by NDPK for DNA repair (). Therefore, CMPK knockdown in serum-deprived cells has an effect on repair through the local change of dCTP at DNA damage site, which is not revealed by the measurement of total dCTP pool. In conclusion, this study brings out 2 notions in the relation of nucleotide supply and DNA repair. First, the nucleotide pool has a determining role in the rate of repair during the cell cycle. Second, the complex formation of nucleotide synthesis enzymes at DNA damage sites compensates the situation when nucleotide supply from cytosol is in short to support an efficient DNA repair.
Materials and Methods
Antibodies and reagents
Anti-R1 (sc-11733, for IF or WB), anti-R2 (sc-10844, for WB), horseradish peroxidase (HRP)-conjugated goat anti-mouse, goat anti-rabbit, donkey anti-goat antibodies were obtained from Santa Cruz. Anti-γH2AX (phospho-Ser139-H2AX) antibodies were obtained from Millipore (05–636, for IF) and GeneTex (GTX127340, for IF). Anti-(6-4) photoproduct (64 M-2, for IF) antibody was obtained from Cosmo Bio Co. Ltd (Tokyo, Japan). Anti-β-tubulin (for WB), anti-M2 (F-3165, for IF or WB), FITC, TRITC-conjugated anti-mouse, Alexa fluor 633, FITC, TRITC-conjugated anti-rabbit antibodies, TRITC-conjugated anti-goat antibody, cytidine and deoxycytidine were obtained from Sigma-Aldrich. Anti-HA (clone 12C5A, for WB) antibody was obtained from Roche. Anti-mCherry antibody (for WB) was obtained from GeneTex. TurboFect transfection reagent was purchased from Thermo Scientific.
Cell lines
293FT cells for lentivirus production were purchased from Invitrogen. 293FT, HEK 293T, HeLa and MCF-7 cells were maintained in DMEM (Invitrogen) containing 10% fetal bovine serum (Invitrogen) at 37°C under 5% CO2. Serum deprivation was performed by maintaining cells in serum-free DMEM for 72 h to decrease cellular dNTP levels.
Plasmids
Human CMPK cDNA clone were obtained from VYM Genome Research Center, Taiwan. The expression vectors of flag-CMPK wild-type and mutant form (ΔN) were constructed by insertion of PCR products from CMPK cDNA at HindIII (5′) and KpnI (3′) sites of pCMV2 vector. The expression vector of mCherry-Nuc-R1C was constructed by insertion of the DNA fragment covering C-terminal 701–792 amino acids of R1 subunit of RNR into BamHI site of mCherry-Nuc vector. pCMVdeltaR8.91, pMD.G, pLKO.1 CMPK, Tip60, R1, LacZ and Luciferase shRNA were obtained from the National RNAi Core Facility, Taiwan. pCDNA3-HA-Tip60 was kindly provided by Dr. Li-Jung, Juan (Genomics Research Center, Academia Sinica, Taiwan).
Lentiviral shRNA production for infection
293FT cells were co-transfected with pCMVdeltaR8.91, pMD.G and pLKO.1 shRNA plasmids. After transfection for 60 h, supernatants containing lentivirus were filtered through PVDF membrane (pore, 0.45 μm, Millipore) and concentrated using Amicon Ultra column (100 kDa molecular weight cut-off, Millipore). Cells were infected by the virus soup containing 8 μg/ml polybrene (Sigma). After 6 h infection, cells were washed twice by PBS and cultured in a complete medium.
dNTP determination
A total of 106 cells were extracted with 1 ml of ice-cold 60% methanol at −20°C overnight, followed by centrifugation at 16,000 g for 30 min. The supernatants were boiled for 3 min and dried under vacuum. The pellets were dissolved in nuclease free water for dNTP measurement based on the method previously described.Citation33
Western blot
Cells were harvested in RIPA buffer (100 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% SDS, 0.2% deoxycholic acid, 0.5% NP-40, 0.5 mM dithiothreitol, 1 mM EDTA pH 8.0), and proteins in each cell lysate were separated by SDS-PAGE and analyzed by immunoblot using specific antibodies and horseradish peroxidase (HRP)-conjugated secondary antibodies. The enhanced chemiluminescent substrate (Millipore) for reaction was added, and the signal was detected by UVP bioimaging system.
Immunofluorescence
The staining of (6-4) photoproducts (6-4 pp) were performed according to manufacture's manual. For γH2AX, M2 or R1 staining, cells were fixed with 4% paraformaldehyde at room temperature for 15 min followed by permeabilization with TBS (50 mM Tris-HCl, pH 7.4, 150 mM NaCl) containing 0.3% Triton X-100 at room temperature for 10 min. For PCNA staining, cells were treated with CSK buffer (10 mM PIPES pH 7.0, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2) containing 0.5% Triton X-100 at room temperature for 1 min followed by fixation with methanol at −20°C for 5 min. After fixation and permeabilization, cells were blocked with 5.5% normal goat serum in TBST (TBS containing 0.1% Triton X-100) or with 5% BSA in TBST, after which cells were stained with primary antibody with the indicated dilution in TBST: 6-4 pp (1: 1500), γH2AX (1: 1000), M2 (1: 5000), R1 (1: 100), PCNA (1: 200) overnight at 4°C. After washing 3 times with TBST (5 min for each time), cells were stained with secondary antibodies and Hoechst 33342 at room temperature for 1 h. Finally, cells were washed 3 times with TBST. After mounting, cells were observed and photographed by using Olympus BX51 microscope. For quantification of immunofluorescence intensity, images acquired from the same exposure setting were analyzed by ImageJ software. The intensities were normalized by the background value for analysis.
UV irradiation
MCF-7 cells grown on coverslips were UVC irradiated by UVP CL-1000 UV crosslinker. For whole-cell irradiation, cells in PBS were subjected to 15 J/M2 of UV irradiation. For localized UV micro-irradiation, polycarbonate isopore membrane with 3 μm pore size filters (Millipore) were placed on top of cells in Hank's balanced salt solution (HBSS), and irradiated with 120 J/M2 UV. Following removal of the filters, the cells were recovered with fresh medium and processed to immunofluorescence assay with specific primary antibodies.
RT-qPCR
Total RNA was extracted by TRIzol reagent (Invitrogen) according to the manufacturer's protocol. RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific) was used for cDNA synthesis. Real-time quantitative PCR analysis was performed using Stepone Real-Time PCR system (Applied Biosystems) and Fast SYBER Green Master Mix (KAPA Biosystems). The expression of Tip60 was normalized to 18S rRNA. The primer sequences were as followed: human Tip60 sense primer 5′- ACCCTGCCTCCCTACCAGCG-3′, antisense primer 5′- ACCCTGCCTCCCTACCAGCG-3′; human 18S rRNA sense primer 5′-CGACGACCCATTCGAACGTCT-3′, antisense primer 5′-CTCTCCGGAATCGAACCCTGA-3′.
Laser micro-irradiation
HeLa cells were seeded at the appropriated density on glass-bottomed dishes. After overnight of incubation, cells were incubated with 2 μg/ml Hochest 33342 for 10 min. Laser micro-irradiation was carried out using a FluoView 1000 confocal microscope (Olympus) with a 405 nm laser diode (fastest mode, SIM scanner, 100 msec). Five min post micro-irradiation, cells were washed with CSK buffer (10 mM PIPES pH 7.0, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2) containing 0.1% Triton X-100 for 5 min prior to fixation with 4% of paraformaldehyde for 15 min then processed to immunofluorescence assay with specific primary antibodies. Cells were analyzed and photographed by using FluoView 1000 confocal microscope.
Immunoprecipitation
HEK 293T cells were transfected with indicated plasmids. After 18 to 24 h of transfection, cells were lysed with RIPA buffer containing with 1 mM PMSF, 10 mM NaF, 2 mM Na3VO4, 1 × protease inhibitor and protein phosphatase inhibitor cocktail (Sigma). Cells lysates were pre-cleared with BSA-blocked Sepharose beads prior to incubation with BSA-blocked M2 beads (Sigma) or HA monoclonal antibody-conjugated beads (Sigma) 1.5 to 2 h at 4°C. The beads were washed 3 times with RIPA buffer and proteins on the beads were boiled at 95°C 10 min with 2 × Laemmli buffer and separated by SDS-PAGE.
987618_Supplementary_Materials.zip
Download Zip (457 KB)Acknowledgments
We thank Dr. Li-Jung, Juan for kindly giving the expression vector of HA-Tip60, Chih-Wei Chen and Ming-Yun Hsieh for the construction of expression vectors of mCherry-NLS-R1C and flag-CMPK, respectively.
Funding
This study was supported by grants, NHRI-EX103–10005NI and MOST 103-2321-B-010-015, from National Health Research Institute and Ministry of Science and Technology, Taiwan (R.O.C.), respectively, and Aim for Top University Plan in National Yang-Ming University sponsored by the Ministry of Education, Taiwan (R.O.C). This study is also supported by NIH grant AI 38204 and Dr. Yung-Chi Cheng is a fellow of National Foundation of Cancer Research.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Imaizumi T, Jean-Louis F, Dubertret ML, Bailly C, Cicurel L, Petchot-Bacqué JP, Dubertret L. Effect of human basic fibroblast growth factor on fibroblast proliferation, cell volume, collagen lattice contraction: in comparison with acidic type. J Dermatol Sci 1996; 11:134-41; PMID:8869034; http://dx.doi.org/10.1016/0923-1811(95)00431-9
- Guarino E, Salguero I, Kearsey SE. Cellular regulation of ribonucleotide reductase in eukaryotes. Semin Cell Dev Biol 2014; 30:97-103; PMID:24704278; http://dx.doi.org/10.1016/j.semcdb.2014.03.030
- Lee MH, Wang L, Chang ZF. The contribution of mitochondrial thymidylate synthesis in preventing the nuclear genome stress. Nucleic Acids Res 2014; 42:4972-84; PMID:24561807; http://dx.doi.org/10.1093/nar/gku152
- Pontarin G, Ferraro P, Bee L, Reichard P, Bianchi V. Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc Natl Acad Sci U S A 2012; 109:13302-7; PMID:22847445; http://dx.doi.org/10.1073/pnas.1211289109
- Chabes AL, Pfleger CM, Kirschner MW, Thelander L. Mouse ribonucleotide reductase R2 protein: a new target for anaphase-promoting complex-Cdh1-mediated proteolysis. Proc Natl Acad Sci U S A 2003; 100:3925-9; PMID:12655059; http://dx.doi.org/10.1073/pnas.0330774100
- Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem 2006; 75:681-706; PMID:16756507; http://dx.doi.org/10.1146/annurev.biochem.75.103004.142443
- D’Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, Saraf A, Florens L, Washburn MP, Pagano M. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 2012; 149:1023-34; http://dx.doi.org/10.1016/j.cell.2012.03.043
- Amiri M, Conserva F, Panayiotou C, Karlsson A, Solaroli N. The human adenylate kinase 9 is a nucleoside mono- and diphosphate kinase. Int J Biochem Cell Biol 2013; 45:925-31; PMID:23416111; http://dx.doi.org/10.1016/j.biocel.2013.02.004
- Brady WA, Kokoris MS, Fitzgibbon M, Black ME. Cloning, characterization and modeling of mouse and human guanylate kinases. J Biol Chem 1996; 271:16734-40; PMID:8663313; http://dx.doi.org/10.1074/jbc.271.19.11427
- Van Rompay AR, Johansson M, Karlsson A. Phosphorylation of deoxycytidine analog monophosphates by UMP-CMP kinase: molecular characterization of the human enzyme. Mol Pharmacol 1999; 56:562-9; PMID:10462544
- Liou JY, Dutschman GE, Lam W, Jiang Z, Cheng YC. Characterization of human UMP/CMP kinase and its phosphorylation of D- and L-form deoxycytidine analogue monophosphates. Cancer Res 2002; 62:1624-31; PMID:11912132
- Fischle W. Tip60-ing the balance in DSB repair. Nat Cell Biol 2009; 11:1279-81; PMID:19884884; http://dx.doi.org/10.1038/ncb1109-1279
- Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A 2005; 102:13182-7; PMID:16141325; http://dx.doi.org/10.1073/pnas.0504211102
- Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, Price BD. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol 2009; 11:1376-82; PMID:19783983; http://dx.doi.org/10.1038/ncb1982
- Niida H, Katsuno Y, Sengoku M, Shimada M, Yukawa M, Ikura M, Ikura T, Kohno K, Shima H, Suzuki H, et al. Essential role of Tip60-dependent recruitment of ribonucleotide reductase at DNA damage sites in DNA repair during G1 phase. Genes Dev 2010; 24:333-8; PMID:20159953; http://dx.doi.org/10.1101/gad.1863810
- Niida H, Shimada M, Murakami H, Nakanishi M. Mechanisms of dNTP supply that play an essential role in maintaining genome integrity in eukaryotic cells. Cancer Sci 2010; 101:2505-9; PMID:20874841; http://dx.doi.org/10.1111/j.1349-7006.2010.01719.x
- Hu CM, Yeh MT, Tsao N, Chen CW, Gao QZ, Chang CY, Lee MH, Fang JM, Sheu SY, Lin CJ, et al. Tumor cells require thymidylate kinase to prevent dUTP incorporation during DNA repair. Cancer Cell 2012; 22:36-50; PMID:22789537; http://dx.doi.org/10.1016/j.ccr.2012.04.038
- Anderson DD, Woeller CF, Chiang EP, Shane B, Stover PJ. Serine hydroxymethyltransferase anchors de novo thymidylate synthesis pathway to nuclear lamina for DNA synthesis. J Biol Chem 2012; 287:7051-62; PMID:22235121; http://dx.doi.org/10.1074/jbc.M111.333120
- Berkovich E, Monnat RJ Jr, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol 2007; 9:683-90; PMID:17486112; http://dx.doi.org/10.1038/ncb1599
- Wakasugi M, Sasaki T, Matsumoto M, Nagaoka M, Inoue K, Inobe M, Horibata K, Tanaka K, Matsunaga T. Nucleotide excision repair-dependent DNA double-strand break formation and ATM signaling activation in mammalian quiescent cells. J Biol Chem 2014; 289:28730-7; PMID:25164823; http://dx.doi.org/10.1074/jbc.M114.589747
- Lima-Bessa KM, Menk CF. Skin cancer: lights on genome lesions. Curr Biol 2005; 15:R58-61; PMID:15668158; http://dx.doi.org/10.1016/j.cub.2004.12.056
- Liu L, Lee J, Zhou P. Navigating the nucleotide excision repair threshold. J Cell Physiol 2010; 224:585-9; PMID:20458729; http://dx.doi.org/10.1002/jcp.22205
- Marti TM, Hefner E, Feeney L, Natale V, Cleaver, JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc Natl Acad Sci U S A 2006; 103:9891-6; PMID:16788066; http://dx.doi.org/10.1073/pnas.0603779103
- Matsumoto M, Yaginuma K, Igarashi A, Imura M, Hasegawa M, Iwabuchi K, Date T, Mori T, Ishizaki K, Yamashita K, et al. Perturbed gap-filling synthesis in nucleotide excision repair causes histone H2AX phosphorylation in human quiescent cells. J Cell Sci 2007; 120:1104-12; PMID:17327276; http://dx.doi.org/10.1242/jcs.03391
- Liou JY, Lai HR, Hsu CH, Chang WL, Hsieh MJ, Huang YC, Cheng, YC. Modulation of human UMP/CMP kinase affects activation and cellular sensitivity of deoxycytidine analogs. Biochem Pharmacol 2010; 79:381-8; PMID:19765547; http://dx.doi.org/10.1016/j.bcp.2009.09.010
- Van Rompay AR, Norda A, Lindén K, Johansson M, Karlsson A. Phosphorylation of uridine and cytidine nucleoside analogs by two human uridine-cytidine kinases. Mol Pharmacol 2001; 59:1181-6; PMID:11306702
- Sabini E, Hazra S, Ort S, Konrad M, Lavie A. Structural basis for substrate promiscuity of dCK. J Mol Biol 2008; 378:607-21; PMID:18377927; http://dx.doi.org/10.1016/j.jmb.2008.02.061
- Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, Nakazawa Y, Niimi A, Miki Y, Jaspers NG, et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol Cell 2010; 37:714-27; PMID:20227374; http://dx.doi.org/10.1016/j.molcel.2010.02.009
- Mourón S, Rodriguez-Acebes S, Martínez-Jiménez MI, García-Gómez S, Chocrón S, Blanco L, Méndez J. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 2013; 20:1383-9; http://dx.doi.org/10.1038/nsmb.2719
- Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Mose S, Prescianotto-Baschong C, Sauer U, Jenoe P, Hall MN. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013; 339:1320-3; PMID:23429704; http://dx.doi.org/10.1126/science.1228771 PMID:
- Ben-Sahra I, Howell JJ, Asara, JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013; 339:1323-8; PMID:23429703; http://dx.doi.org/10.1126/science.1228792
- Xu Y, Johansson M, Karlsson A. Human UMP-CMP kinase 2, a novel nucleoside monophosphate kinase localized in mitochondria. J Biol Chem 2008; 283:1563-71; PMID:17999954; http://dx.doi.org/10.1074/jbc.M707997200
- Ferraro P, Franzolin E, Pontarin G, Reichard P, Bianchi V. Quantitation of cellular deoxynucleoside triphosphates. Nucleic Acids Res 2010; 38:e85; PMID:20008099; http://dx.doi.org/10.1093/nar/gkp1141