
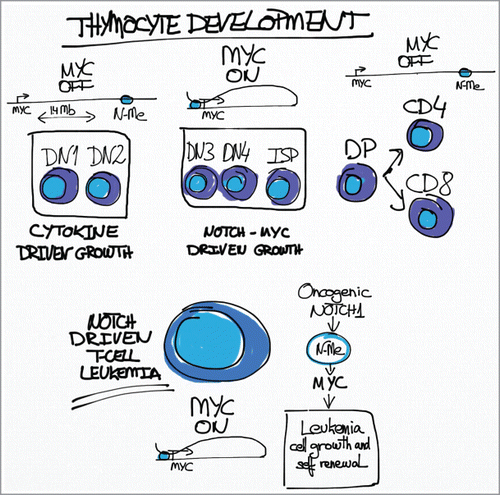
Constitutive activation of NOTCH signaling plays a central role in the pathogenesis of over 60% of T-cell acute lymphoblastic leukemias (T-ALL) harboring activating mutations in NOTCH1 or disruption of FBXW7, a critical negative regulator of NOTCH signaling.Citation1 Expression of oncogenic NOTCH1 drives T-cell transformation activating a transcriptional program that includes multiple genes promoting leukemia cell growth and proliferation. Among these, the MYC oncogene has been highlighted as an important mediator of NOTCH1 induced transformation as NOTCH1 signaling induces transcriptional upregulation of MYC expression and NOTCH1 and MYC share multiple common target genes involved in leukemia cell growth.Citation2 Still, demonstrating direct regulation of MYC by NOTCH1 has remained elusive as NOTCH1 fails to activate MYC promoter activity in reporter assays. This enigmatic link between NOTCH1 and MYC in T-ALL has been now clarified with the identification of N-Me (for NOTCH MYC enhancer), a long-range distal enhancer controlling MYC expression and T-cell transformation downstream of NOTCH1 ().Citation3 The N-Me enhancer is located +1,427 kb from MYC in a large gene desert region extending telomeric from the MYC locus and represents the strongest peak of NOTCH1 binding in ChIP-seq analysis of T-ALL cells. Notably, and despite its distal genomic location, this regulatory element directly interacts with and controls MYC promoter activity as demonstrated by chromatin configuration 3C analysis and reporter assays. The functional importance of N-Me in the control of MYC expression is further supported by multispecies DNA sequence homology analyses which revealed remarkable conservation of this region in mammals, birds and reptiles. In addition, analysis of functional chromatin marks showed bona fide active enhancer features including P300 occupancy, high levels of H3K4me1 and low levels of H3K4me3 associated with N-Me. Moreover, detailed epigenetic profiling across 64 haematopoietic and non-haematopoietic cell lines and tissues revealed that this regulatory element is located within a major super-enhancer specifically active in T-cells. Consistently with these results, analysis of N-Me knockout and conditional knockout mice demonstrated a critical role for this long-range enhancer in T-cell development and NOTCH1-induced T-ALL.Citation3 Thus, N-Me deficient mice showed marked thymic atrophy with depletion of CD4 CD8 double positive thymocytes. This phenotype was cell autonomous and was associated with decreased Myc expression in DN3, DN4 and ISP T-cell progenitors. Importantly, these T-cell developmental defects recapitulate those observed in T-cell specific Myc knockout mice,Citation4 and were rescued by retroviral expression of MYC, formally demonstrating a strict requirement for N-Me mediated Myc expression in T-cell development. In addition, the NOTCH-N-Me-Myc regulatory axis showed to be essential for NOTCH1 induced T-ALL. Thus, N-Me knockout bone marrow progenitor cells failed to generate leukemia upon retroviral expression of oncogenic NOTCH1, and secondary deletion of N-Me in fully established NOTCH1-induced T-ALL resulted in profound antileukemic effects and almost complete suppression of leukemia initiating cell activity. Strikingly, loss of one copy of N-Me showed intermediate phenotypes including partial reduction of thymocyte numbers; reduced penetrance of T-ALL upon oncogenic NOTCH1 activation; and delayed tumor progression of fully established NOTCH1-driven tumors. These results highlight the exact dose-dependent function of the NOTCH1-NMe-Myc regulatory circuitry. Finally, genomic profiling of human leukemias identified recurrent focal duplications encompassing the N-Me enhancer in T-ALL supporting that increased N-Me dosage may contribute to T-cell transformation by strengthening NOTCH1 driven MYC expression.
Overall our results highlight the importance of long-range enhancers involved in the control of MYC expression in the pathogenesis of human cancer and are in line with previous results in myeloid leukemia and colon cancer.Citation5,6 Still, the detailed characterization of the N-Me enhancer opens new outstanding questions that remain to be addressed. Most intriguingly, NOTCH1 and MYC play important developmental roles in multiple tissues other than T-cells and NOTCH signaling regulation of MYC expression has been implicated in the pathogenesis of breast cancer and other solid tumors;Citation7,8 however N-Me seems to be strictly T-cell specific, suggesting that other as yet unidentified NOTCH-controlled MYC enhancers may be responsible for MYC expression outside T-cells. In addition, it is unclear what factors drive the T-cell specific activity of the N-Me enhancer.
Finally, it has not escaped our attention that our identification of N-Me duplications in T-ALL support a potentially important and broader role for chromosomal copy number alterations and mutations occurring in intergenic regulatory elements in the pathogenesis of human cancer.
Figure 1. Schematic representation of N-Me activity during thymocyte development and in NOTCH1-induced T-ALL.
References
- Tzoneva G, Ferrando AA. Recent advances on NOTCH signaling in T-ALL. Curr Top Microbiol Immunol 2012; 360:163-82; PMID:22673746; http://dx.doi.org/10.1007/82_2012_232
- Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, Barnes KC, O'Neil J, Neuberg D, Weng AP, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A 2006; 103:18261-6; PMID:17114293, http://dx.doi.org/10.1073/pnas.0606108103
- Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, Xu L, Castillo-Martin M, Llobet-Navás D, Cordon-Cardo C, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med 2014; 20:1130-7); PMID:25194570, http://dx.doi.org/10.1038/nm.3665
- Dose, M., Khan I, Guo Z, Kovalovsky D, Krueger A, von Boehmer H, Khazaie K, Gounari F. c-Myc mediates pre-TCR-induced proliferation but not developmental progression. Blood 2006; 108:2669-77; PMID:16788099, http://dx.doi.org/10.1182/blood-2006-02-005900
- Shi, J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe JS, Minder JL, Mercan F, Wang E, Eckersley-Maslin MA, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev 2013; 27:2648-62; PMID:24285714, http://dx.doi.org/10.1101/gad.232710.113
- Sur, IK, Hallikas O, Vähärautio A, Yan J, Turunen M, Enge M, Taipale M, Karhu A, Aaltonen LA, Taipale J. Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science 2012; 338:1360-3; PMID:23118011, http://dx.doi.org/10.1126/science.1228606
- Allen TD, Rodriguez EM, Jones KD, Bishop JM. Activated Notch1 induces lung adenomas in mice and cooperates with Myc in the generation of lung adenocarcinoma. Cancer Res 2011; 71, 6010-8; PMID:21803744, http://dx.doi.org/10.1158/0008-5472.CAN-11-0595
- Klinakis A., Szabolcs M, Politi K, Kiaris H, Artavanis-Tsakonas S, Efstratiadis A. Myc is a Notch1 transcriptional target and a requisite for Notch1-induced mammary tumorigenesis in mice. Proc Natl Acad Sci U S A 2006; 103, 9262-7; PMID:16751266, http://dx.doi.org/10.1073/pnas.0603371103