
Cancer arises as a consequence of somatic mutations and clonal amplification of tumor cells. However, it has been known since many years that cancer cells also accumulate a multitude of epigenetic alterations including aberrant DNA methylation.Citation1 Interestingly, two converse trends of DNA methylation changes were observed in many tumors. On the one hand, promoters of tumor suppressor genes are often hypermethylated leading to the silencing of the genes. On the other hand, a global DNA hypomethylation including heterochromatic repetitive elements is observed which leads to genomic instability. In a recent publication in Cell Cycle, Wu et al.Citation2 for the first time present a mechanism that could explain both of these inverse changes by a mutation in DNMT1, an important DNA methyltransferase in human cells.
In mammals, DNA methylation mainly takes place at CpG sites, where methylation occurs on both DNA strands and it cycles between full methylation and hemimethylation after DNA replication. DNMT1 has a preference for the methylation of hemimethylated CpG sites and it was initially believed to purely function as maintenance methyltransferase, which copies DNA methylation patterns after replication. However, it became clear that DNA methylation is dynamically regulated and the control of DNA methyltransferases including DNMT1 plays a central role in the generation and maintenance of DNA methylation patterns.Citation3
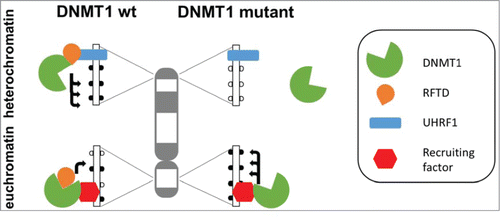
DNMT1 is a large multidomain protein. Its C-terminal catalytic domain is under tight allosteric control by several N-terminal domains, including the RFTS domain (RFTD), which has 3 roles in the regulation of DNMT1:Citation2,4-7 First, it blocks the catalytic pocket and by this it reduces the catalytic activity of DNMT1. Second, it interacts with other proteins including UHRF1, which can target DNMT1 to replication foci (via its interaction with hemimethylated DNA), but also to heterochromatin (via its interaction with H3K9 methylated histones). Third, interaction with UHRF1 can relieve the RFTD mediated allosteric repression of DNMT1 activity. Wu et al. now show that deletion of the RFTS domain leads to converse changes of DNA methylation which mimic the alterations observed in cancer cells (). One the one hand, the activity of DNMT1 is increased leading to DNA hypermethylation at sites, where DNMT1 is targeted in an RFTD independent manner, for example at promoter region of tumor suppressor genes. One the other hand, deletion of RFTD disrupts the interaction of DNMT1 and UHRF1 leading to the loss of heterochromatic localization DNMT1 and hypomethylation of heterochromatic SAT2 elements. It is conceivable to speculate that somatic DNMT1 mutations in cancer cells which disrupt folding of the RFTS domain could have similar effects. Therefore, the paper by Wu et al. is an important contribution to our understanding of the causes of the complex DNA methylation changes observed in cancer cells.
Figure 1. Schematic picture of the changes in DNA methylation caused by the loss-of-function of the RFTS domain (RFTD) in DNMT1. Wildtype DNMT1 is recruited by an interaction of UHRF1 with RFTD to heterochromatin. At the same time, the interaction of UHRF1 with RFTD relieves the inhibition of DNMT1 caused by binding of RFTD to the catalytic pocket leading to strong methylation of heterochromatic repeats. In the DNMT1 mutant which lost the RFTD this targeting does not occur and the methylation of the heterochromatic DNA is reduced. At the same time DNMT1 is recruited by other factors to euchromatic elements like promoters in an RFTD independent fashion. Here, the activity of wildtype DNMT1 is reduced by the RFTS domain and the DNMT1 mutant without RFTD is more active, leading to higher methylation of such regions.
References
- Baylin SB, Jones PA. Nat Rev Cancer 2011; 11(10):726-34; PMID:21941284; http://dx.doi.org/10.1038/nrc3130
- Wu B-K, et al. Cell Cycle 2014; 13(20):3222-31; PMID:16403455; http://dx.doi.org/10.4161/15384101.2014.950886
- Jeltsch A, Jurkowska RZ. TIBS 2014; 39(7):310-8; PMID:24947342
- Syeda F, et al. J Biol Chem 2011; 286(17):15344-51; PMID:21389349; http://dx.doi.org/10.1074/jbc.M110.209882
- Bashtrykov P, et al. J Biol Chem 2014; 289(7):4106-15; PMID:24368767; http://dx.doi.org/10.1074/jbc.M113.528893
- Bashtrykov P, et al. Chem Bio Chem 2014; 15(5):743-8; PMID:24532244; http://dx.doi.org/10.1002/cbic.201300740
- Berkyurek AC, et al. J Biol Chem 2014; 289(1):379-86; PMID:24253042; http://dx.doi.org/10.1074/jbc.M113.523209