
Abstract
Chemical RNA modifications are present in all kingdoms of life and many of these post-transcriptional modifications are conserved throughout evolution. However, most of the research has been performed on single cell organisms, whereas little is known about how RNA modifications contribute to the development of metazoans. In recent years, the identification of RNA modification genes in genome wide association studies (GWAS) has sparked new interest in previously neglected genes. In this review, we summarize recent findings that connect RNA modification defects and phenotypes in higher eukaryotes. Furthermore, we discuss the implications of aberrant tRNA modification in various human diseases including metabolic defects, mitochondrial dysfunctions, neurological disorders, and cancer. As the molecular mechanisms of these diseases are being elucidated, we will gain first insights into the functions of RNA modifications in higher eukaryotes and finally understand their roles during development.
Introduction
Chemical RNA modifications are a universal phenomenon of molecular biology and occur in all 3 kingdoms of life.Citation1 Virtually every class of RNA molecules in the cell can be post-transcriptionally modified, and certain modifications are evolutionarily conserved and may be indispensable for translation.Citation2 This apparent conservation has benefited our understanding of RNA modification pathways tremendously, because it allowed for the use of simple model organisms like yeast and bacteria. However, this powerful approach has left us with a puzzle. Why are so many RNA modifications evolutionarily conserved, but show no obvious phenotypes in single cell organisms when grown under laboratory conditions?
This apparent lack of phenotypes in yeast has discouraged further studies in higher eukaryotes, preventing us from obtaining a realistic view of the function of RNA modifying pathways in developing organisms. However, it is worth taking a closer look. For example, deleting the ELP1 gene, which is required for 5-methoxycarbonylmethyluridine (mcm5U) and 5-carbamoylmethyluridine (ncm5U) formation at tRNA wobble uridines (U34), leads to subtle phenotypes in yeast.Citation3 In contrast, deleting Ikbkap, the ELP1 homolog in mice, results in early embryonic lethality, revealing an essential role of these modifications in higher eukaryotes.Citation4 Furthermore, the prolific use of next generation sequencing and its application in genome wide association studies (GWAS) have unexpectedly linked several RNA modification genes to human diseases. While this has brought RNA modifications to center stage in certain fields, we still know very little about RNA modifying enzymes in higher eukaryotes.
In this review, we discuss recent findings that link RNA modifications to phenotypes in higher eukaryotes and consider their implication in human disease. Importantly, we will not cover capping, adenylation, deadenylation and editing, but will instead primarily focus on chemical modification of tRNA, mRNA, and to a certain degree, rRNA.
Phenotypes linked to mutations in RNA modifying enzymes have been described in many multicellular organisms (). Their analyses are complicated by several factors: First, functional orthologues of known RNA modifying enzymes have not been identified in all species.Citation5 Second, the modification status of many RNA molecules has been characterized only in a few metazoans,Citation6 and we often rely on analogies to yeast. Finally, phenotypes are complex and diverse. For an overview of phenotypes linked to RNA modification deficiencies, please refer to Table 1 for humans, Table 2 for mice, and Table 3 for zebrafish and flies.
Figure 1. tRNA modification defects and phenotypes in higher eukaryotes. Schematic representation of a tRNA. Modified nucleosides that have been linked to phenotypes in higher eukaryotes are indicated as red circles. The color inside the circle denotes the type of defect observed. Chemical modifications and their causative genes (in brackets) are linked to the respective nucleoside. Gray or black residues depict nucleosides that are either unmodified or not linked to phenotypes. Abbreviations of the nucleosides follow the nomenclature of Modomics (http://modomics.genesilico.pl/).
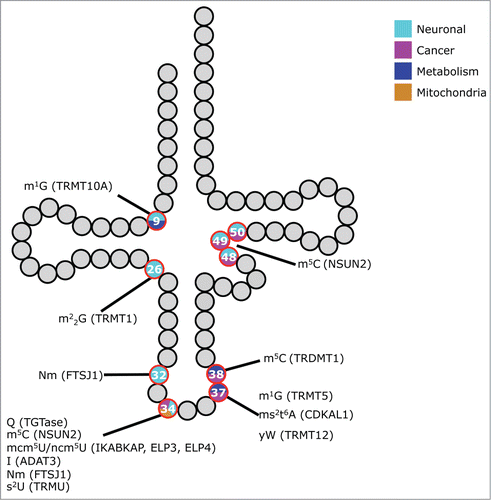
However, common themes emerge from these studies, and it is possible to separate the defects into 4 classes: i) perturbed metabolic pathways, ii) mitochondrial defects, iii) neuronal disorders, and iv) increased susceptibility to cancer. It will become apparent that this classification is somewhat artificial. Certain genes fall into more than one class and it may be a matter of preference, which one is emphasized. Furthermore, complex phenotypes can obscure classification, as it is sometimes difficult to distinguish between primary and secondary defects. Nevertheless, these categories can act as a good starting point to address the complexity.
Metabolic Defects
Metabolic defects combine changes in cellular or organismal metabolism, which lead to altered metabolite levels in the blood of patients. In the last decades, the spread of a high sugar and high fat diet has lead to an epidemic of metabolic disorders. In particular, type 2 diabetes (TIID) receives a lot of attention since it affects health and economies on a global scale.Citation7
Several modification genes have been linked to metabolic defects, including the fat mass and obesity associated (FTO) gene, CDK5 regulatory subunit associated protein 1-like 1 (CDKAL1), tRNA aspartic acid methyltransferase 1 (TRDMT1), and tRNA methyltransferase 10 homolog A (TRMT10A). FTO, initially called fatso, was identified in a mouse mutant characterized by fused toes and defects in brain development and body axis control.Citation8 However, FTO immediately entered the limelight when independent GWAS linked single nucleotide polymorphisms (SNPs) in intron 1 of FTO to TIID and obesity.Citation9-12 The causative SNPs are strongly associated with early onset obesity and are predictive for average weight differences of 3 kg.Citation9,11,13 FTO is a 2-oxogluterate dependent oxygenase and can demethylate 6-methyladenosine (m6A) or form N6-hydroxymethyladenosine (hm6A) and N6-formyladenosine (f6A) in mRNA.Citation14-17
The tight regulation of m6A methylation in mRNA is crucial during gametogenesis in Drosophila and mice and embryogenesis in Arabidopsis.Citation18,19 In flies and plants the gene is also essential later during development.Citation19,20 Thus, it is conceivable that FTO is critical in regulating mRNA by modulating their modification, which may change mRNA turnover and translation and its interaction with RNA binding proteins. For an in-depth review, please refer to. Citation21
FTO is expressed in many human tissues. Expression levels in the hypothalamus are very high, which is consistent with a role in energy regulationCitation9,14 Mice with a FTO knockout die soon after birth as a result of a growth retardation that starts at day 2.Citation22,23 Both lean mass and fat mass are reduced in the neuron-specific or full knockout strains, while oxygen consumption and CO2 production increase.Citation22,23 Interestingly, a dominant missense mutant, which reduces FTO activity to ∼30%, develops a similar phenotype.Citation24 Complementary results stem from mouse models that overexpress additional cDNA copies of FTO, leading to a dose dependent increase of body weight.Citation25
Analyses of the m6A pattern in cellular transcripts identified thousands of potential mRNA targetsCitation26-28 and different mechanisms downstream of m6A methylation have been proposed, including dopamine receptor response and Wnt signaling.Citation28,29 Knockdown of FTO in zebrafish reduces brain and eye size and perturbs neural crest cell migration and cilia formation.Citation29 This is consistent with pathologies reported in humans. While certain SNPs were found to affect brain size of healthy individuals with no apparent damage,Citation30 other reports link FTO variants to mental retardation and microcephaly.Citation31,32
Recently, the direct role of FTO in obesity was questioned and explained by a long-range interaction of FTO intron 1 with the promoter of the homeobox gene IRX3.Citation33 Importantly, the authors found no correlation between FTO expression levels and obesity. Even though very compelling, the described mechanism does not fully explain the phenotypes of the dominant mouse mutant and of the cDNA overexpression in mice.Citation24,25
CDKAL1 was linked to TIID and obesity at the same time as FTO.Citation10,34-36 It is a methylthiotransferase, required for the generation of 2-methylthio-N6-threonylcarbamoyladenosine (ms2t6A) at position 37 in the anticodon of cytoplasmic tRNALys(UUU).Citation37,38 Proinsulin processing is perturbed in CDKAL1 knockout mice, leading to ER stress and reduced insulin secretion in β-cells.Citation38,39 Reporter assays suggest that the insulin secretion defect is caused by misreading of Lysine-codons in proinsulin.Citation38 Human carriers of TIID alleles show reduced levels of ms2t6A37, they fail to generate and to secrete insulin, and they may respond differently to pharmacological treatment of TIID.Citation40-44 Consistent with this, CDKAL1 alleles correlate strongly with an increased risk for coronary artery diseases, most likely as a downstream effect of TIID.Citation45 Furthermore, CDKAL1 was identified as a risk factor for Crohn's disease (CD) and for Psoriasis. However, the alleles associated with CD and Psoriasis are not linked to TIID.Citation46,47
TRDMT1 causes more subtle phenotypes than FTO or CDKAL1. The protein was described as a DNA methylase but is in fact required to form 5-methylcytidine (m5C) on tRNA and mRNA.Citation48-53 Knockouts in Drosophila, Arabidopsis and mice were indistinguishable from wild type.Citation48,49 However, TRDMT1 may influence cellular metabolism in unexpected ways. Studies in humans link a variant in TRDMT1 to increased folate levels, which may subsequently lower the risk of developing congenital heart diseases or spina bifida during pregnancy.Citation54,55 Furthermore, morpholino-mediated downregulation of TRDMT1 in zebrafish led to malformed eyes, as well as brain and liver defects.Citation50 Whether the more severe phenotype is explained by different requirements in zebrafish or by off-target effects needs to be seen. However, m5C levels are important since the knockout of NOP2/Sun RNA methyltransferase family, member 2 (NSUN2) is characterized by a significant weight reduction.Citation56 Furthermore, the double knockout of TRDMT1 and NSUN2 in mice is synthetic lethal.Citation53 The double mutants are smaller at birth and die within the first days due to a feeding defect. They also exhibit severe skeletal and brain defects, as well as perturbed fat metabolism.Citation53 Interestingly, mouse embryonic fibroblasts derived from the double mutant accumulate tRNA fragments and show signs of reduced protein translation.Citation53 Similarly, TRDMT1 mutant flies contain stress induced tRNA fragments and constitutively upregulate the innate immune system.Citation57,58
Mitochondrial Phenotypes
Mitochondria utilize a minimal set of 22 tRNAs to decode all 60 sense codons.Citation59 To achieve this, only 4 post-transcriptional modifications are required at the wobble position of 10 mitochondrial tRNAs: 5-formylcytidine (f5C), 5-taurinomethyluridine (τm5U), 5-taurinomethyl-2-thiouridine (τm5s2U), and queuosine (Q).Citation60 In such a fine-tuned system, incomplete wobble base modification disrupts protein synthesis and causes errors that can potentiate to mitochondrial dysfunction. Cells with mitochondrial defects generally display significantly reduced oxygen consumption and mitochondrial protein synthesis, as well as reduced ATP production, decreased mitochondrial membrane potential, and increased superoxide levels.Citation61,62
A single heteroplasmic A to G replacement at nucleotide position (np) 3243 in the gene encoding for mt tRNALeu(UAA) is associated with diverse pathological phenotypes, including ocular myopathy, diabetes with associated deafness, and the full mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) syndrome.Citation61,63 Roughly 80% of all MELAS patients carry this transition whereas in approx. Ten% of the cases a T to C transition occurs at np 3271. Neither of the mutations lies at the anticodon of the tRNA. However, both mutations lead to τm5U hypomodification of mt tRNALeu(UAA) that manifests in identical clinical symptoms.Citation63,64 A to G transition at np 8344 of the mt tRNALys(UUU) gene leads to loss of τm5s2U modification at the wobble uridine. Patients with this deficiency suffer from mitochondrial encephalomyopathies, myoclonus epilepsy associated with ragged-red fibers (MERRF) syndrome, which constitutes another subgroup of mitochondrial encephalomyopathia that is clinically distinct from MELAS.Citation60,65
The point mutations associated with MELAS and MERRF function as negative determinants of τm5U biosynthesis by disrupting the tertiary structure of the tRNA,Citation64 thereby preventing its proper recognition by a currently unknown taurine transferase. GTPBP3, the human homolog of Mto1p/Mss1p that synthesizes 5-carboxymethyluridine (cmnm5U) in yeast, is a putative candidate but conclusive evidence is lacking.Citation66
Loss of the τm5U modification in MELAS patients prevents the mt tRNALeu(UAA) from reading UUG codons, whereas no apparent reduction in decoding capability is observed for UUA codons.Citation67 Decoding dysfunctions are exacerbated by the point mutations in MERRF, where τm5s2U hypomodification completely abrogates the ability of mt tRNALys(UUU) to read the cognate codons AAA and AAG. This defect is attributed to the complete loss of s2U modification, a critical factor for stabilizing the codon-anticodon interactions in the ribosome.Citation60,67 A natural suppressor tRNA in MELAS cells can largely rescue the decoding disorder. A second mutation from G to A at np 12300 changes the anticodon sequence of mt tRNALeu(UAG), allowing it to read the same codons as mt tRNALeu(UAA).Citation60,61,68,69 Surprisingly, this mutant tRNA can be τm5U modified despite a mere 42% sequence homology to mt tRNALeu(UAA).Citation69 Acquisition of the wobble modification in another isoacceptor tRNA is key to suppressing MELAS and highlights the importance of translational control through nucleoside modifications.
NSUN4 performs m5C methylation of C911 in mitochondrial 12S rRNA.Citation70 Both NSUN4 and its cofactor MTERF4 are essential and their deletion leads to early embryonic lethality in mice.Citation70,71 A heart specific knockout is viable but significantly shortens the lifespan. Mitochondrial translation fails despite a significant increase of transcript levels, giving rise to nonfunctional mitochondria and the ultimately lethal phenotype.Citation70,71
Neurodegenerative and Neurological Defects
Several neurological defects are linked to the absence of homologues of the ELP complex, which is required for the formation of mcm5U34 and ncm5U34 in 11 cytoplasmic tRNAs in different organisms.Citation72-74 The best understood case is familial dysautonomia (FD), also termed the Riley-Day syndrome.Citation75 This devastating autosomal recessive disorder severely affects neurons of the autonomic nervous system, resulting in the absence of sensory neurons. The lack of autonomic control ultimately leads to premature death mainly through cardiovascular failure or respiratory defects.Citation76 99.5% of the disease alleles affect a donor splice site in the IKBKAP gene and lead to tissue specific skipping of exon 20 and introduction of a premature stop codon, thereby drastically reducing IKAP protein levels in neurons.Citation77-79 However, in non-neuronal tissues the remaining wild-type transcripts suffice to ensure cell survival.Citation79 Importantly, reduced levels of IKAP lead to reduced levels of xm5U in cells of FD patients.80 Since FD is caused by a tissue specific splicing defect, attempts to find compounds that may correct IKBKAP splicing and relieve some of the symptoms have been undertaken.Citation81-84 Several small molecules have been identified, but only Kinetin has shown positive clinical effects.Citation85 Thus, FD is the only RNA modification disease for which treatment options may exist through restoration of RNA modification levels.
However, why does FD affect only sensory neurons? Are they particularly vulnerable to RNA modification defects, or are they targeted by a tissue specific splicing defect? Answers to these questions may come from animal models. The full knockout of ikbkap leads to lethality around embryonic day 12.5.Citation4 Developing embryos are small and show defects in extraembryonic tissues as well as neuronal and vascular development.Citation4 When a deletion allele of exon 20 is combined with a full knockout allele, the phenotypes are similar to the full knockout, suggesting that the mutant protein is non-functional.Citation86 A mouse model that recapitulates FD symptoms combines the exon 20 deletion with a hypomorphic allele that reduces IKAP activity to 10%.Citation87 These mutants are significantly smaller at birth and frequently die thereafter due to a feeding defect, reminiscent of TRDMT1/NSUN2 mutant mice.Citation53,87 Furthermore, the animals exhibit gastro-intestinal defects and reduction of sympathetic ganglia and other phenotypes characteristic of FD.Citation87 Interestingly, a tissue specific knockout in testis triggers meiotic defects and subsequent apoptosis of germ cells,Citation74 showing that IKAP function is essential also in non-neuronal tissues. In summary, FD is caused by reduced IKAP function in a dose dependent manner. The enzymatic nature of the ELP complex and the long half-life of tRNAs may generate sufficient levels of modified tRNA to allow for survival even at low expression levels. Thus, the reason why sensory neurons are affected may be a combination of neurons being more sensitive to perturbations together with a tissue specific splicing defect that reduces IKAP levels more in neuronal tissues than in other cells.
Not only IKAP, but almost every subunit of the ELP complex has been linked to a disease. ELP2 is a risk gene for intellectual disabilities (ID),Citation88 while ELP3 is associated with non-familial Amyotrophic lateral scelerosis (ALS).Citation89 Similarly, Drosophila mutants of ELP3 show neuronal defects and their motor neurons branch abnormally.Citation89 Axon length of motor neurons in zebrafish is decreased in a dose dependent manner upon knockdown of elp3,Citation89 and elpc-1 and elpc-3 mutants exhibit learning defects in Caenorhabditis elegans.Citation90 In humans, Rolandic epilepsy, a generally mild form of childhood epilepsy, has been associated with ELP4.Citation91,92 However, it is unclear whether ELP4 is the causal gene. Potentially, the neighboring PAX6 is controlled by regulatory elements in the introns of ELP4, as proposed for FTO and IRX3.Citation33,93 Poly, the Drosophila homolog of ELP6, is essential in larval development and its absence causes major metabolic changes and brain defects.Citation94 While the ELP complex is linked to multiple diseases, no pathologies have been associated with the URM1 pathway, which is required for s2U formation on 3 mcm5-modified tRNA.Citation95 However, in Arabidopsis, mutants of this pathway affect root growth.Citation96
FtsJ RNA methyltransferase homolog 1 (FTSJ1), a homolog of Trm7, is required for the 2’-O-ribose methylation at G34, U34, C34 and C32 of several tRNA.Citation97,98 It was linked to non-syndromic X-linked ID but also to general mental performance.Citation99-105 Most of the mutations described so far result in a loss-of-function of the protein, either by impairing the critical SAM-binding domain of the protein or by degradation of the mutant mRNA.Citation99,101 There are no morphological brain phenotypes in patients.Citation99 However, high expression levels in the fetal brain may reflect the importance of the gene during brain development.Citation99 Interestingly, 2 studies report that duplications of a genomic region that contains FTSJ1 lead to ID accompanied by mild dysmorphic features.Citation105,106 The fact that both duplication and loss of function leads to similar phenotypes may suggest that FTSJ1 levels need to be precisely controlled. To better understand this seemingly paradoxical situation, a systematic manipulation of FTSJ1 expression levels in animal models is of utmost significance. However, these models are currently not available.
Mutations in NSUN2 are linked to ID and a Dubowitz-like syndrome, characterized by mild microcephaly and congenital heart defects.Citation107-110 Similarly, perturbed NSUN2 function results in deficits in short-term memory and behavioral assays in mice and flies,Citation107 as well as reduced fertility in flies, mice and humans.Citation107,109,111 Expression levels in the brain are high in mice and flies, consistent with the phenotypes.Citation107,109,112 NSUN2 catalyzes the formation of m5C at the wobble position 34 of tRNALeu(CAA), as well as positions 48-50 of several tRNA molecules. However, m5C sites are also found in other ncRNA and mRNA, where they are enriched in the UTR and overlap with Argonaute binding sites.Citation52,113 Thus, most models to explain the phenotypes imply changes of RNA turnover in response to methylation of mRNA or non-coding RNAs.Citation113,114 A different mechanism implies tRNA as mediators of phenotypes: Interestingly, 5’ tRNA-fragments accumulate in tissues and cells in NSUN2 knockout mice and both NSUN2 and the tRNA-fragments co-localize with cellular stress markers.Citation115 This model postulates that angiogenin, a cellular ribonuclease, generates these fragments in response to cellular stress, thereby downregulating translation rates.Citation115 However, it will be critical to dissect the contribution of each of the proposed mechanisms to understand the phenotypes.
SNPs in TRMT10A, the mammalian homolog of Trm10, which modifies 12 tRNA species by 1-methylguanosine (m1G) at position 9 in yeast, were reported to cause ID, microcephaly and perturbed glucose metabolism.Citation115,116 Inactivation of TRMT10A by siRNA in rat β-cells sensitizes the cells against free fatty acid induced ER stress and induces apoptosis.Citation117
Internal m6A methylation of mRNA in humans is achieved by a methylase-complex, that consist of at least 3 subunits: METTL3, a methyltransferase acts in concert with it homolog METTL14 and Wilms’ tumor 1-associating protein (WTAP).Citation118-120 WTAP targets the other subunits to mRNA but is enzymatically inactive.Citation119 While METTL3 and METTL14 methylate their substrates independently in vitro, their activity is strongly enhanced when acting cooperatively.Citation118 Inactivation of METTL3 or WTAP in zebrafish causes severe developmental defects characterized by smaller heads and eyes and a curved notochord accompanied by increased apoptosis.Citation119 Interestingly, stem cells loose their ability to maintain a pluripotent state, when METTL3 or METTL14 are downregulated.Citation120 Consistently, knockout mice die during early embryonic development,Citation121,122 and cells of the inner cell mass fail to differentiate into mesoderm or endoderm.Citation122
Malformations of the zebrafish brain are also observed in morpholino-mediated knockdowns of the 3 conserved small nucleolar RNAs (snoRNA) U26, U44 and U78, suggesting that translation defects consistently affect brain development.Citation123
Finally, 2 more genes are known to cause ID: mutations in ADAT3, which is required for the formation of inosine (I34) at the anticodon of several tRNAs, cause severe ID and strabismus,Citation124 and TRMT1, the homolog of Trm1p, is required for the formation of N2,N2-dimethylguanosine (m22G).Citation88 However, experiments to decipher these phenotypes are still pending.
Cancer
Cancer is the collective term for a broad group of complex diseases characterized by unregulated cell growth resulting in malignant tumors. Defects in tRNA modification have been directly tied to cell proliferation and malignancy for a number of lymphomas,Citation125 leukemiasCitation125,126 and carcinomas, including skin, breast, bladder, and colorectal cancers.Citation127-131 In the late 1960s Borek and co-workers isolated tRNA from various tumor tissues and reported elevated levels of methylated nucleosides, as well as an increased tRNA methylase activity in the cells. This suggests that tRNA hypermodification might have a specific functional role in tumor cells (reviewed in Citation132). However, several subsequent studies have shown that tumor-specific tRNA species tend to be hypomodified rather than hypermodified.Citation133
Insufficient queuosine Q modification in tRNA from neoplastic cells is a well-studied example of tRNA hypomodification.Citation125,135 Queuine, a derivative of 7-aminomethyl-7-deazaguanine, is found exclusively in the first position of the tRNA anticodon in the form of Q, mannosyl-queuosine (manQ), and galactosyl-queuosine (galQ).Citation135,136 Eukaryotes are unable to synthesize queuine and rely on a salvage system that acquires it as a nutrient factor. Q containing tRNAs are fully modified under normal physiological conditions, but are often hypomodified in undifferentiated rapidly growing cells, embryonic tissues, and neoplastically-transformed cells. Q deficiency is associated with lymphoma, leukemia and various kinds of tumors.Citation125,137,138 In fast growing tumor cells queuine is replaced by guanine at the tRNA wobble position.Citation134 This observation cannot be fully explained by insufficient queuine uptake, since addition of excess amounts of exogenous queuine only partially restored Q modification to tumor tRNA.Citation139 Furthermore, slowly growing tumors are also deficient in Q modification.Citation133 This suggests that Q hypomodification arises either due to functional impairment of the tRNA-guanine transglycosylase (TGTase) enzyme and subsequent lack of queuine incorporation into tRNA, or due to a queuine salvage pathway deficiency, or a combination of both.Citation135,136
The physiological role of queuine remains ill defined. More recently Q modification has been implicated in protecting the cellular antioxidant defense system during malignancy.Citation140 It has also been suggested that replacement of queuine with guanine in the anticodon has the potential to alter mRNA codon recognition and, as a result, gene expression.Citation126,141 Treatment of a human promyelocytic leukemia (HL-60) cell line with 6-thioguanine, a guanine analog, resulted in growth inhibition and cell differentiation.Citation126 This has been attributed to altered codon recognition by tRNAs containing the guanine analogs, allowing read-through of amber (UAG) stop codons in the presence of guanine in the wobble position of tRNATyr.Citation126,141
Although tRNA molecules possess a considerable number of nucleoside modifications, only a handful of mammalian tRNA methyltransferases have been described to date. These include the 6 putative families of m5C methyltransferases of which only NSUN2 and TRDMT1 have been characterized, as well as other tRNA methyltransferases including tRNA methyltransferase homolog 12 (TRMT12),Citation127 TRMT10A,Citation131 and human AlkB homolog 8 (ALKBH8).Citation130 Aberrant function of these methyltransferases has been linked to tumor formation. In addition, several other RNA modification enzymes have been associated with cancers in humans (please refer to for a comprehensive overview).
Figure 2. RNA modification genes associated with human malignancies. Schematic representation of various cancers for which increase (red) or decrease (blue) of tRNA modification gene copy number, or expression level, has been reported. (Source: Cosmic and Oncomine).
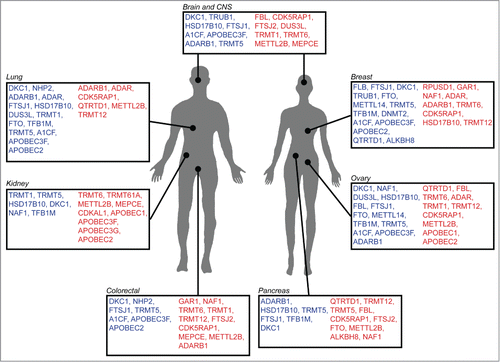
NSUN2 is a direct target gene of c-Myc, a well-known proto-oncogene that triggers proliferation and decrease cell adhesion. Significant upregulation of NSUN2 expression was detected both in primary tumors and metastases of breast carcinomas and in malignant squamous cell carcinomas, whereas benign papillomas showed slightly lower expression levels. Knockdown of NSUN2 expression by RNAi inhibits tumor growth in squamous cell carcinoma xenografts in a dose-dependent manner,Citation142 making it a potential target for cancer therapy. Interestingly, the cancer drug Azacytidine, which inhibits DNA methylation, reduces m5C levels in RNA in tissue culture cells, suggesting that RNA demethylation may contribute to the efficacy of Azacytidine.Citation143
Correct function of the human tRNA-(N1G37) methyltransferase (TRMT5), which catalyzes methyl transfer from S-adenosyl methionine (AdoMet) to guanine to synthesize m1G at position 37 in many tRNAs, is a critical determinant in preventing ribosomal frameshifts.Citation144 TRMT5 expression is significantly downregulated in several colorectal cancers whereas overexpression occurs in head and neck cancers (Oncomine). Another modification occurring at position 37 of tRNAPhe is the formation of wybutosine by TRMT12, whose expression is significantly upregulated in several breast cancer cell linesCitation127 and is also prevalent in leukemia, but tends to be downregulated in colorectal cancers (Oncomine).
Weak Phenotypes
Mice deleted for ALKBH8, which is required for the last step of mcm5U synthesis in cytoplasmic tRNA, appear normal up to 20 months of age.Citation145 However, tRNASec, which is required for the recoding of UGA stop-codons to selenocysteine, lacks mcm5U and 5-methoxycarbonylmethyl-2’-O-methyluridine (mcm5Um),Citation145 leading to reduced levels of the selenoprotein glutathione peroxidase 1 (Gpx1), as well as glutathione peroxidase activity.Citation145 Nevertheless, this deficiency does not cause apparent morphological phenotypes.
Like for ALKBH8, knockout mice of ALKBH5 - a sister enzyme of FTO - overall seem anatomically normal.Citation146 However, males have small testes with perturbed morphology and a reduced number of dysfunctional spermatozoa. It is unclear whether the localization of ALKBH5 to nuclear speckles, and its effects for the recruitment of certain RNA processing factors and mRNA, contribute to this phenotype.Citation146
Discussion
We observe diverse phenotypes when RNA modification genes are perturbed in higher eukaryotes, but we are only starting to grasp the complexity of these pathways. It appears relatively straightforward to rationalize that mitochondrial dysfunction severely affects different tissues. However, to connect other deficiencies to the observed pathologies is much more challenging. In yeast, certain modification defects can be rescued by overexpressing their respective modification target.Citation3,98,147 However, similar experiments have not been performed in metazoans. Hence, it needs to be shown that these phenotypes are actually caused by RNA modification defects.
Why do phenotypes differ between single cell and multicellular organisms? Yeasts and bacteria are generally grown under favorable conditions in the lab and have been selected for rapid growth under this regime for thousands of generations. Thus, they can by no means be compared to the complexity of a developing organism that coordinates multiple differentiation events in parallel. Despite a high degree of buffering and compensatory mechanisms, seemingly subtle but systematic perturbations may trigger processes that will ultimately affect cellular and organismal function.
It will be interesting albeit challenging to understand phenotypes that were identified in GWAS. Even in the well-defined case of IKAP, several attempts were necessary to generate a mouse model that recapitulates FD, suggesting that the relevant phenotype is not a full knockout. Thus, the generation of hypomorphic mutants or introduction of relevant SNPs into model organisms may be required to generate realistic disease models. Furthermore, the ratio of cognate versus near-cognate tRNA genes can potentially affect the interference by unmodified tRNAs.Citation148 Finally, tissue specific RNA expression or different genetic mouse backgrounds can affect the outcome of a phenotype significantly (please refer also to the checklist in Box 1).Citation149,150 All these factors need to be kept in mind, when modeling RNA modification pathologies, and it is possible that certain defects in humans can be best mimicked by studying inducible pluripotent stem cells (iPSC) and differentiated cells derived from them.
Table 4. Factors to consider:
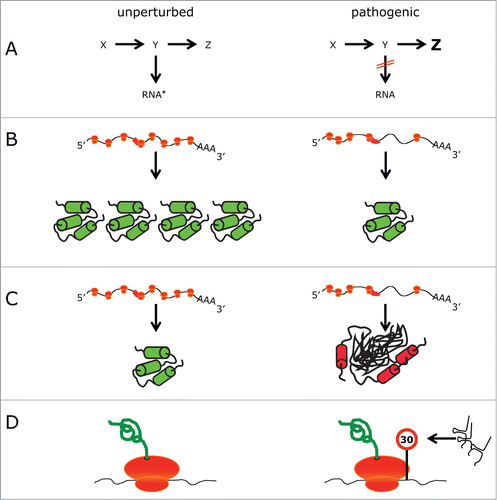
What are the potential mechanisms by which RNA modifications may affect multicellular species? First, perturbation of RNA modification pathways may reroute certain metabolites, thereby affecting other pathways and causing toxicity (). Second, translation is likely to be affected in many modification defects of tRNA, rRNA or mRNA. However, direct evidence in vivo is scarce. Codon specific translation defects may lead to decreased production of individual proteins that are required either in certain cell types or under specific stress conditions (). Third, codon specific translation defects may give rise to protein aggregates and subsequent failure of protein homeostasis (S.L unpublished results; ). Finally, the occurrence of tRNA fragments in a number of defects is an interesting observation.Citation53,115 In this scenario, 5’ halves of tRNA reduce cellular translation rates, which perturbs the cell (). Similarly, accumulation of tRNA fragments was also reported in patients suffering from neurodegeneration and microcephaly upon loss of tRNA kinase CLP1 activity.Citation151,152 However, this effect may not be independent of protein aggregation and further analysis of the causal relationship between these phenomena will be imperative. At this point it is conceivable that tRNA fragments are both a causal factor as well as a downstream marker of cellular defects. Nevertheless, translational modulation is a likely mechanism underlying many pathologies, since a large number of mutations affect the tRNA anticodon and, in particular, the wobble position ().
Figure 3. Models of how RNA modification defects cause phenotypes. Comparison of different scenarios in an unperturbed (left) and pathogenic (right) situation. (A) A metabolic pathway is blocked, leading to the absence of modified RNA (indicated by an asterisk) and the build up of a different metabolite (in this case Z). (B) Ribosomes (orange) translating an mRNA, which contains a region that is difficult to translate (red box). In the pathogenic situation translation is perturbed, leading to a lower amount of protein. (C) As in (B). Perturbed translation prevents the folding of some proteins into their native state resulting in perturbed protein homeostasis. (D) tRNA fragments cause a slowdown of translation.
Finally, why do certain alleles occur at such high frequency in the population? For example, certain FTO variants account for adverse phenotypes, such as predisposition to obesityCitation9-12 and reduced brain volume,Citation30 while the same allele protects against depressionCitation153 and the risk for alcohol dependence.Citation154 It is therefore difficult to decide whether a specific allele is problematic or not. This Janus faced character of certain alleles may explain their high prevalence in the population.
We live and perform research in exciting times. Unbiased approaches identify new links between hitherto “boring” genes and new phenotypes. At the same time the role of classical genes like FTO is questioned. It will require an enormous effort to combine new animal models and iPSC with state of the art biochemistry and molecular biology. Only when we look systematically, we will be able to understand why certain mutants “modify or die” and others don’t!
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Table 1. RNA modification genes associated with disease in humans
Table 2. Mouse models for RNA modification genes.
Table 3. Drosophila and zebrafish models for RNA modification genes.
Acknowledgments
The authors wish to thank Brigitte Saß for proofreading of the manuscript.
Funding
This work was supported by grants from the Max Planck Society, the North Rhine-Westphalian Ministry for Innovation, Science and Research [314-400 010 09], and the European Research Council [ERC-2012-StG 310489-tRNAmodi] to S.A.L. L.P.S. is a Sigrid Jusélius Fellow (2012-2014).
References
- Grosjean H. Nucleic acids are not boring long polymers of only four types of nucleotides: a guided tour. In: Grosjean H, ed. DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution. 2009. 1-18.
- Grosjean H, Breton M, Sirand-Pugnet P, Tardy F, Thiaucourt F, Citti C, Barré A, Yoshizawa S, Fourmy D, de Crécy-Lagard V, et al. Predicting the inimal translation apparatus: lessons from the reductive evolution of mollicutes. PLoS Genet 2014; 10:e1004363; PMID:24809820
- Esberg A, Huang B, Johansson MJO, Byström AS. Elevated levels of two tRNA species bypass the requirement for elongator complex in transcription and exocytosis. Mol Cell 2006; 24:139-48; PMID:17018299[http://dx.doi.org/10.1016/j.molcel.2006.07.031]
- Chen Y-T, Hims MM, Shetty RS, Mull J, Liu L, Leyne M, Slaugenhaupt SA. Loss of mouse Ikbkap, a subunit of elongator, leads to transcriptional deficits and embryonic lethality that can be rescued by human IKBKAP. Mol Cell Biol 2009; 29:736-44; PMID:19015235[http://dx.doi.org/10.1128/MCB.01313-08]
- Towns WL, Begley TJ. Transfer RNA methytransferases and their corresponding modifications in budding yeast and humans: activities, predications, and potential roles in human health. DNA Cell Biol 2012; 31:434; PMID:22191691[http://dx.doi.org/10.1089/dna.2011.1437]
- Suzuki T, Suzuki T. A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res 2014; 42:7346-57; PMID:24831542[http://dx.doi.org/10.1093/nar/gku390]
- Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature 2001; 414:782-7; PMID:11742409[http://dx.doi.org/10.1038/414782a]
- van der Hoeven F, Schimmang T, Volkmann A, Mattei MG, Kyewski B, Rüther U. Programmed cell death is affected in the novel mouse mutant Fused toes (Ft). Development 1994; 120:2601-7; PMID:7956835
- Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JRB, Elliott KS, Lango H, Rayner NW, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007; 316:889-94; PMID: 17434869[http://dx.doi.org/ 10.1126/science.1141634]
- Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, et al. A Genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007; 316:1341-5; PMID:17463248[http://dx.doi.org/10.1126/science.1142382]
- Dina C, Meyre D, Gallina S, Durand E, Körner A, Jacobson P, Carlsson LMS, Kiess W, Vatin V, Lecoeur C, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet 2007; 39:724-6; PMID:17496892[http://dx.doi.org/10.1038/ng2048]
- Scuteri A, Sanna S, Chen W-M, Uda M, Albai G, Strait J, Najjar S, Nagaraja R, Orrú M, Usala G, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet 2007; 3:e115; PMID:17658951
- Liu C, Mou S, Cai Y. FTO gene variant and risk of overweight and obesity among children and adolescents: a systematic review and meta-analysis. PLoS One 2013; 8:e82133; PMID:24278475; http://dx.doi.org/10.1371/journal.pone.0082133
- Gerken T, Girard CA, Tung Y-CL, Webby CJ, Saudek V, Hewitson KS, Yeo GSH, McDonough MA, Cunliffe S, McNeill LA, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 2007; 318:1469-72; PMID:17991826[http://dx.doi.org/10.1126/science.1151710]
- Jia G, Yang C-G, Yang S, Jian X, Yi C, Zhou Z, He C. Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett 2008; 582:3313-9; PMID:18775698[http://dx.doi.org/10.1016/j.febslet.2008.08.019]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang Y-G, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 2011; 7:885-7; PMID:22002720[http://dx.doi.org/10.1038/nchembio.687]
- Fu Y, Jia G, Pang X, Wang RN, Wang X, Li CJ, Smemo S, Dai Q, Bailey KA, Nóbrega MA, et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat Commun 2013; 4:1798; PMID:23653210[http://dx.doi.org/10.1038/ncomms2822]
- Zhong S, Li H, Bodi Z, Button J, Vespa L, Herzog M, Fray RG. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 2008; 20:1278-88; PMID:18505803[http://dx.doi.org/10.1105/tpc.108.058883]
- Hongay CF, Orr-Weaver TL. Drosophila Inducer of MEiosis 4 (IME4) is required for Notch signaling during oogenesis. Proc Natl Acad Sci USA 2011; 108:14855-60.
- Bodi Z, Zhong S, Mehra S, Song J, Graham N, Li H, May S, Fray RG. Adenosine methylation in arabidopsis mRNA is associated with the 3′ end and reduced levels cause developmental defects. Front Plant Sci 2012; 3:48; PMID:22639649[http://dx.doi.org/10.3389/fpls.2012.00048]
- Sibbritt T, Patel HR, Preiss T. Mapping and significance of the mRNA methylome. WIREs RNA 2013; 4:397-422; PMID: 23681756[http://dx.doi.org/ 10.1002/wrna.1166]
- Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Brüning JC, Rüther U. Inactivation of the Fto gene protects from obesity. Nature 2009; 458:894-8; PMID:19234441[http://dx.doi.org/10.1038/nature07848]
- Gao X, Shin Y-H, Li M, Wang F, Tong Q, Zhang P. The fat mass and obesity associated gene FTO functions in the brain to regulate postnatal growth in mice. PLoS One 2010; 5:e14005; PMID:21103374; http://dx.doi.org/10.1371/journal.pone.0014005
- Church C, Lee S, Bagg EAL, McTaggart JS, Deacon R, Gerken T, Lee A, Moir L, Mecinović J, Quwailid MM, et al. A mouse model for the metabolic effects of the human fat mass and obesity associated FTO gene. PLoS Genet 2009; 5:e1000599. Available from: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=19680540&retmode=ref&cmd=prlinks; PMID:19680540; http://dx.doi.org/10.1371/journal.pgen.1000599
- Church C, Moir L, McMurray F, Girard C, Banks GT, Teboul L, Wells S, Brüning JC, Nolan PM, Ashcroft FM, et al. Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet 2010; 42:1086-92; PMID :21076408[http://dx.doi.org/ 10.1038/ng.713]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 2012; 149:1635-46; PMID:22608085[http://dx.doi.org/10.1016/j.cell.2012.05.003]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012; 485:201-6; PMID:22575960[http://dx.doi.org/10.1038/nature11112]
- Hess ME, Hess S, Meyer KD, Verhagen LAW, Koch L, Brönneke HS, Dietrich MO, Jordan SD, Saletore Y, Elemento O, et al. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci 2013; 16:1042-8; PMID:23817550; http://dx.doi.org/10.1038/nn.3449
- Osborn DPS, Roccasecca RM, McMurray F, Hernandez-Hernandez V, Mukherjee S, Barroso I, Stemple D, Cox R, Beales PL, Christou-Savina S. Loss of FTO antagonises Wnt signaling and leads to developmental defects associated with ciliopathies. PLoS One 2014; 9:e87662; PMID:24503721
- Ho AJ, Stein JL, Hua X, Lee S, Hibar DP, Leow AD, Dinov ID, Toga AW, Saykin AJ, Shen L, et al. A commonly carried allele of the obesity-related FTO gene is associated with reduced brain volume in the healthy elderly. Proc Natl Acad Sci USA 2010; 107:8404-9.
- Boissel S, Reish O, Proulx K, Kawagoe-Takaki H, Sedgwick B, Yeo GSH, Meyre D, Golzio C, Molinari F, Kadhom N, et al. Loss-of-function mutation in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. Am J Hum Genet 2009; 85:106-11; PMID: 19559399[http://dx.doi.org/ 10.1016/j.ajhg.2009.06.002]
- van den Berg L, de Waal HD-V, Han JC, Ylstra B, Eijk P, Nesterova M, Heutink P, Stratakis CA. Investigation of a patient with a partial trisomy 16q including the fat mass and obesity associated gene (FTO): fine mapping and FTO gene expression study. Am J Med Genet A 2010; 152A:630-7; PMID:20186806[http://dx.doi.org/10.1002/ajmg.a.33229]
- Smemo S, Tena JJ, Kim K-H, Gamazon ER, Sakabe NJ, Gómez-Marín C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 2014; 507:371-5; PMID:24646999[http://dx.doi.org/10.1038/nature13138]
- Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, Walters GB, Styrkarsdottir U, Gretarsdottir S, Emilsson V, Ghosh S, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet 2007; 39:770-5; PMID:17460697[http://dx.doi.org/10.1038/ng2043]
- Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research, Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PIW, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007; 316:1331-6.
- Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JRB, Rayner NW, Freathy RM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336-41; PMID:17463249[http://dx.doi.org/ 10.1126/science.1142364]
- Arragain S, Handelman SK, Forouhar F, Wei F-Y, Tomizawa K, Hunt JF, Douki T, Fontecave M, Mulliez E, Atta M. Identification of eukaryotic and prokaryotic methylthiotransferase for biosynthesis of 2-methylthio-N6-threonylcarbamoyladenosine in tRNA. J Biol Chem 2010; 285:28425-33; PMID:20584901[http://dx.doi.org/10.1074/jbc.M110.106831]
- Wei F-Y, Suzuki T, Watanabe S, Kimura S, Kaitsuka T, Fujimura A, Matsui H, Atta M, Michiue H, Fontecave M, et al. Deficit of tRNA(Lys) modification by Cdkal1 causes the development of type 2 diabetes in mice. J Clin Invest 2011; 121:3598-608; PMID:21841312[http://dx.doi.org/10.1172/JCI58056]
- Ohara-Imaizumi M, Yoshida M, Aoyagi K, Saito T, Okamura T, Takenaka H, Akimoto Y, Nakamichi Y, Takanashi-Yanobu R, Nishiwaki C, et al. Deletion of CDKAL1 Affects Mitochondrial ATP Generation and First-Phase Insulin Exocytosis. PLoS ONE 2010; 5:e15553; PMID:21151568
- Kirchhoff K, Machicao F, Haupt A, Schäfer SA, Tschritter O, Staiger H, Stefan N, Häring H-U, Fritsche A. Polymorphisms in the TCF7L2, CDKAL1 and SLC30A8 genes are associated with impaired proinsulin conversion. Diabetologia 2008; 51:597-601; PMID:18264689[http://dx.doi.org/10.1007/s00125-008-0926-y]
- Stančáková A, Pihlajamäki J, Kuusisto J, Stefan N, Fritsche A, Häring H, Andreozzi F, Succurro E, Sesti G, Boesgaard TW, et al. Single-nucleotide polymorphism rs7754840 of CDKAL1Is associated with impaired insulin secretion in nondiabetic offspring of type 2 diabetic subjects and in a large sample of men with normal glucose tolerance. J Clin Endocrinol Metab 2008; 93:1924-30; PMID:18285412[http://dx.doi.org/10.1210/jc.2007-2218]
- Groenewoud MJ, Dekker JM, Fritsche A, Reiling E, Nijpels G, Heine RJ, Maassen JA, Machicao F, Schäfer SA, Häring H-U, et al. Variants of CDKAL1 and IGF2BP2 affect first-phase insulin secretion during hyperglycaemic clamps. Diabetologia 2008; 51:1659-63; PMID:18618095[http://dx.doi.org/10.1007/s00125-008-1083-z]
- Xie P, Wei F-Y, Hirata S, Kaitsuka T, Suzuki T, Suzuki T, Tomizawa K. Quantitative PCR measurement of tRNA 2-methylthio modification for assessing type 2 diabetes risk. Clin Chem 2013;; 59:1604-12. Available from: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=23974085&retmode=ref&cmd=prlinks; PMID:23974085; http://dx.doi.org/10.1373/clinchem.2013.210401
- Schroner Z, Javorský M, Halušková J, Klimčáková L, Babjaková E, Fabianová M, Slabá E, Kozárová M, Tkáč I. Variation in CDKAL1 gene is associated with therapeutic response to sulphonylureas. Physiol Res 2012; 61:177-83; PMID:22292718
- Saade S, Cazier J-B, Ghassibe-Sabbagh M, Youhanna S, Badro DA, Kamatani Y, Hager J, Yeretzian JS, El-Khazen G, Haber M, et al. Large scale association analysis identifies three susceptibility loci for coronary artery disease. PLoS One 2011; 6:e29427; PMID: 22216278;http://dx.doi.org/10.1371/journal.pone.0029427
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet 2008; 40:955-62; PMID: 18587394[http://dx.doi.org/ 10.1038/ng.175]
- Quaranta M, Burden AD, Griffiths CEM, Worthington J, Barker JN, Trembath RC, Capon F. Differential contribution of CDKAL1 variants to psoriasis, Crohn's disease and type II diabetes. Genes Immun 2009; 10:654-8; PMID: 19587699[http://dx.doi.org/ 10.1038/gene.2009.51]
- Kunert N, Marhold J, Stanke J, Stach D, Lyko F. A Dnmt2-like protein mediates DNA methylation in Drosophila. Development 2003; 130:5083-90; PMID:12944428[http://dx.doi.org/10.1242/dev.00716]
- Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh C-L, Zhang X, Golic KG, Jacobsen SE, Bestor TH. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006; 311:395-8; PMID:16424344[http://dx.doi.org/10.1126/science.1120976]
- Rai K, Chidester S, Zavala CV, Manos EJ, James SR, Karpf AR, Jones DA, Cairns BR. Dnmt2 functions in the cytoplasm to promote liver, brain, and retina development in zebrafish. Genes Dev 2007; 21:261-6; PMID:17289917[http://dx.doi.org/10.1101/gad.1472907]
- Schaefer M, Pollex T, Hanna K, Lyko F. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res 2009; 37:e12; PMID:19059995
- Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, Suter CM, Preiss T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res 2012; 40:5023-33; PMID:22344696; http://dx.doi.org/10.1093/nar/gks144
- Tuorto F, Liebers R, Musch T, Schaefer M, Hofmann S, Kellner S, Frye M, Helm M, Stoecklin G, Lyko F. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat Struct Mol Biol 2012; 19:900-5; PMID:22885326[http://dx.doi.org/10.1038/nsmb.2357]
- Franke B, Vermeulen SHHM, Steegers-Theunissen RPM, Coenen MJ, Schijvenaars MMVAP, Scheffer H, Heijer den M, Blom HJ. An association study of 45 folate-related genes in spina bifida: Involvement of cubilin (CUBN) and tRNA aspartic acid methyltransferase 1(TRDMT1). Birth Defect Res A 2009; 85:216-26; PMID:19161160
- Chowdhury S, Hobbs CA, MacLeod SL, Cleves MA, Melnyk S, James SJ, Hu P, Erickson SW. Molecular genetics and metabolism. Mol Gen Meta 2012; 107:596-604; PMID:23059056[http://dx.doi.org/10.1016/j.ymgme.2012.09.022]
- Blanco S, Kurowski A, Nichols J, Watt FM, Benitah SA, Frye M. The RNA–methyltransferase misu (NSun2) poises epidermal stem cells to differentiate. PLoS Genet 2011; 7:e1002403; PMID:22144916; http://dx.doi.org/10.1371/journal.pgen.1002403
- Durdevic Z, Hanna K, Gold B, Pollex T, Cherry S, Lyko F, Schaefer M. Efficient RNA virus control in Drosophila requires the RNA methyltransferase Dnmt2. EMBO Rep 2013; 14:269-75; PMID:23370384[http://dx.doi.org/10.1038/embor.2013.3]
- Durdevic Z, Mobin MB, Hanna K, Lyko F, Schaefer M. The RNA methyltransferase Dnmt2 is required for efficient Dicer-2-dependent siRNA pathway activity in Drosophila. Cell Rep 2013; 4:931-7; PMID:24012760; http://dx.doi.org/10.1016/j.celrep.2013.07.046
- Watanabe K. Unique features of animal mitochondrial translation systems. Proc Japan Acad 2010; 86:11-39; PMID:20075606
- Suzuki T, Nagao A, Suzuki T. Human mitochondrial diseases caused by lack of taurine modification in mitochondrial tRNAs. WIREs RNA 2011; 2:376-86; PMID:21957023[http://dx.doi.org/10.1002/wrna.65]
- Meziane El A, Lehtinen SK, Hance N, Nijtmans LG, Dunbar D, Holt IJ, Jacobs HT. A tRNA suppressor mutation in human mitochondria. Nat Genet 1998; 18:350-3; PMID:9537417[http://dx.doi.org/10.1038/ng0498-350]
- Kirino Y, Suzuki T. Human mitochondrial diseases associated with tRNA wobble modification deficiency. RNA Biol 2005; 2:41-4; PMID:17132941[http://dx.doi.org/10.4161/rna.2.2.1610]
- Yasukawa T, Suzuki T, Ueda T, Ohta S, Watanabe K. Modification defect at anticodon wobble nucleotide of mitochondrial tRNAs(Leu)(UUR) with pathogenic mutations of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. J Biol Chem 2000; 275:4251-7; PMID:10660592[http://dx.doi.org/10.1074/jbc.275.6.4251]
- Kirino Y, Goto Y, Campos Y, Arenas J, Suzuki T. Specific correlation between the wobble modification deficiency in mutant tRNAs and the clinical features of a human mitochondrial disease. Proc Natl Acad Sci USA 2005; 102:7127-32.
- Yasukawa T, Suzuki T, Ishii N, Ueda T, Ohta S, Watanabe K. Defect in modification at the anticodon wobble nucleotide of mitochondrial tRNA(Lys) with the MERRF encephalomyopathy pathogenic mutation. FEBS Lett 2000; 467:175-8; PMID:10675533[http://dx.doi.org/10.1016/S0014-5793(00)01145-5]
- Villarroya M, Prado S, Esteve JM, Soriano MA, Aguado C, Perez-Martinez D, Martinez-Ferrandis JI, Yim L, Victor VM, Cebolla E, et al. Characterization of human GTPBP3, a GTP-binding protein involved in mitochondrial tRNA modification. Mol Cell Biol 2008; 28:7514-31; PMID:18852288[http://dx.doi.org/10.1128/MCB.00946-08]
- Kirino Y, Yasukawa T, Ohta S, Akira S, Ishihara K, Watanabe K, Suzuki T. Codon-specific translational defect caused by a wobble modification deficiency in mutant tRNA from a human mitochondrial disease. Proc Natl Acad Sci USA 2004; 101:15070-5.
- Meziane El A, Lehtinen SK, Holt IJ, Jacobs HT. Mitochondrial tRNALeu isoforms in lung carcinoma cybrid cells containing the np 3243 mtDNA mutation. Hum Mol Genet 1998; 7:2141-7; PMID:9817933[http://dx.doi.org/10.1093/hmg/7.13.2141]
- Kirino Y, Yasukawa T, Marjavaara SK, Jacobs HT, Holt IJ, Watanabe K, Suzuki T. Acquisition of the wobble modification in mitochondrial tRNALeu(CUN) bearing the G12300A mutation suppresses the MELAS molecular defect. Hum Mol Genet 2006; 15:897-904; PMID:16446307
- Metodiev MD, Spåhr H, Loguercio Polosa P, Meharg C, Becker C, Altmueller J, Habermann B, Larsson N-G, Ruzzenente B. NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. PLoS Genet 2014; 10:e1004110; PMID:24516400
- Cámara Y, Asin-Cayuela J, Park CB, Metodiev MD, Shi Y, Ruzzenente B, Kukat C, Habermann B, Wibom R, Hultenby K, et al. MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab 2011; 13:527-39; PMID:21531335[http://dx.doi.org/10.1016/j.cmet.2011.04.002]
- Huang B, Johansson MJO, Byström AS. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA 2005; 11:424-36; PMID:15769872[http://dx.doi.org/10.1261/rna.7247705]
- Mehlgarten C, Jablonowski D, Wrackmeyer U, Tschitschmann S, Sondermann D, Jager G, Gong Z, Byström AS, Schaffrath R, Breunig KD. Elongator function in tRNA wobble uridine modification is conserved between yeast and plants. Mol Microbiol 2010; 76:1082-94; PMID:20398216[http://dx.doi.org/10.1111/j.1365-2958.2010.07163.x]
- Lin F-J, Shen L, Jang C-W, Falnes PØ, Zhang Y. Ikbkap/Elp1 deficiency causes male infertility by disrupting meiotic progression. PLoS Genet 2013; 9:e1003516; PMID:23717213
- Riley CM, Day RL. Central autonomic dysfunction with defective lacrimation; report of five cases. Pediatrics 1949; 3:468-78; PMID:18118947
- Gold-von Simson G, Axelrod FB. Familial dysautonomia: update and recent advances. Curr Probl Pediatr Adolesc Health Care 2006; 36:218-37; PMID:16777588[http://dx.doi.org/10.1016/j.cppeds.2005.12.001]
- Slaugenhaupt SA, Blumenfeld A, Gill SP, Leyne M, Mull J, Cuajungco MP, Liebert CB, Chadwick B, Idelson M, Reznik L, et al. Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am J Hum Genet 2001; 68:598-605; PMID:11179008[http://dx.doi.org/10.1086/318810]
- Anderson SL, Coli R, Daly IW, Kichula EA, Rork MJ, Volpi SA, Ekstein J, Rubin BY. Familial dysautonomia is caused by mutations of the IKAP gene. Am J Hum Genet 2001; 68:753-8; PMID:11179021[http://dx.doi.org/10.1086/318808]
- Cuajungco MP, Leyne M, Mull J, Gill SP, Lu W, Zagzag D, Axelrod FB, Maayan C, Gusella JF, Slaugenhaupt SA. Tissue-specific reduction in splicing efficiency of IKBKAP due to the major mutation associated with familial dysautonomia. Am J Hum Genet 2003; 72:749-58; PMID:12577200[http://dx.doi.org/10.1086/368263]
- Karlsborn T, Tükenmez H, Chen C, Byström AS. Biochemical and Biophysical Research Communications. Biochem Biophys Res Commun 2014; 1-5.
- Anderson SL, Qiu J, Rubin BY. EGCG corrects aberrant splicing of IKAP mRNA in cells from patients with familial dysautonomia. Biochem Biophys Res Commun 2003; 310:627-33; PMID:14521957[http://dx.doi.org/10.1016/j.bbrc.2003.09.019]
- Anderson SL, Qiu J, Rubin BY. Tocotrienols induce IKBKAP expression: a possible therapy for familial dysautonomia. Biochem Biophys Res Commun 2003; 306:303-9; PMID:12788105[http://dx.doi.org/10.1016/S0006-291X(03)00971-9]
- Slaugenhaupt SA, Mull J, Leyne M, Cuajungco MP, Gill SP, Hims MM, Quintero F, Axelrod FB, Gusella JF. Rescue of a human mRNA splicing defect by the plant cytokinin kinetin. Hum Mol Genet 2004; 13:429-36; PMID:14709595[http://dx.doi.org/10.1093/hmg/ddh046]
- Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 2009; 461:402-6; PMID:19693009[http://dx.doi.org/10.1038/nature08320]
- Axelrod FB, Liebes L, Gold-von Simson G, Mendoza S, Mull J, Leyne M, Norcliffe-Kaufmann L, Kaufmann H, Slaugenhaupt SA. Kinetin improves IKBKAP mRNA splicing in patients with familial dysautonomia. Pediatr Res 2011; 70:480-3; PMID:21775922[http://dx.doi.org/10.1203/PDR.0b013e31822e1825]
- Dietrich P, Yue J, E S, Dragatsis I. Deletion of exon 20 of the familial dysautonomia gene Ikbkap in mice causes developmental delay, cardiovascular defects, and early embryonic lethality. PLoS One 2011; 6:e27015; PMID:22046433
- Dietrich P, Alli S, Shanmugasundaram R, Dragatsis I. IKAP expression levels modulate disease severity in a mouse model of familial dysautonomia. Hum Mol Genet 2012; 21:5078-90; PMID: 22922231[http://dx.doi.org/10.1093/hmg/dds354]
- Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011; 478:57-63; PMID:21937992[http://dx.doi.org/10.1038/nature10423]
- Simpson CL, Lemmens R, Miskiewicz K, Broom WJ, Hansen VK, van Vught PWJ, Landers JE, Sapp P, Van Den Bosch L, Knight J, et al. Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum Mol Genet 2009; 18:472-81; PMID:18996918[http://dx.doi.org/10.1093/hmg/ddn375]
- Chen C, Tuck S, Byström AS. Defects in tRNA modification associated with neurological and developmental dysfunctions in Caenorhabditis elegans elongator mutants. PLoS Genet 2009; 5:e1000561; PMID:19593383; http://dx.doi.org/10.1371/journal.pgen.1000561
- Strug LJ, Clarke T, Chiang T, Chien M, Baskurt Z, Li W, Dorfman R, Bali B, Wirrell E, Kugler SL, et al. Centrotemporal sharp wave EEG trait in rolandic epilepsy maps to Elongator Protein Complex 4 (ELP4). Eur J Hum Genet 2009; 17:1171-81; PMID:19172991[http://dx.doi.org/10.1038/ejhg.2008.267]
- Strug LJ, Hodge SE, Chiang T, Pal DK, Corey PN, Rohde C. A pure likelihood approach to the analysis of genetic association data: an alternative to Bayesian and frequentist analysis. Eur J Hum Genet 2010; 18:933-41; PMID:20424645[http://dx.doi.org/10.1038/ejhg.2010.47]
- Griffin C, Kleinjan DA, Doe B, van Heyningen V. New 3' elements control Pax6 expression in the developing pretectum, neural retina and olfactory region. Mech Dev 2002; 112:89-100; PMID:11850181[http://dx.doi.org/10.1016/S0925-4773(01)00646-3]
- Bolukbasi E, Vass S, Cobbe N, Nelson B, Simossis V, Dunbar DR, Heck MMS. Drosophila poly suggests a novel role for the Elongator complex in insulin receptor-target of rapamycin signalling. Open Biol 2012; 2:110031; PMID:22645656[http://dx.doi.org/10.1098/rsob.110031]
- Pedrioli PGA, Leidel S, Hofmann K. Urm1 at the crossroad of modifications. “Protein Modifications: Beyond the Usual Suspects” Review Series. EMBO Rep 2008; 9:1196-202; PMID:19047990
- Leiber R-M, John F, Verhertbruggen Y, Diet A, Knox JP, Ringli C. The TOR pathway modulates the structure of cell walls in Arabidopsis. Plant Cell 2010; 22:1898-908; PMID:20530756[http://dx.doi.org/10.1105/tpc.109.073007]
- Pintard L, Lecointe F, Bujnicki JM, Bonnerot C, Grosjean H, Lapeyre B. Trm7p catalyses the formation of two 2′-O-methylriboses in yeast tRNA anticodon loop. EMBO J 2002; 21:1811-20; PMID:11927565[http://dx.doi.org/10.1093/emboj/21.7.1811]
- Guy MP, Podyma BM, Preston MA, Shaheen HH, Krivos KL, Limbach PA, Hopper AK, Phizicky EM. Yeast Trm7 interacts with distinct proteins for critical modifications of the tRNAPhe anticodon loop. RNA 2012; 18:1921-33; PMID:22912484[http://dx.doi.org/10.1261/rna.035287.112]
- Freude K, Hoffmann K, Jensen L, Delatycki M, Desportes V, Moser B, Hamel B, Vanbokhoven H, Moraine C, Fryns J. Mutations in the FTSJ1 gene coding for a novel S-Adenosylmethionine–binding protein cause nonsyndromic x-linked mental retardation. Am J Hum Gen 2004; 75:305-9; PMID:15162322[http://dx.doi.org/10.1086/422507]
- Froyen G, Bauters M, Boyle J, Esch H, Govaerts K, Bokhoven H, Ropers H-H, Moraine C, Chelly J, Fryns J-P, et al. Loss of SLC38A5 and FTSJ1 at Xp11.23 in three brothers with non-syndromic mental retardation due to a microdeletion in an unstable genomic region. Hum Genet 2007; 121:539-47; PMID:17333282[http://dx.doi.org/10.1007/s00439-007-0343-1]
- Takano K, Nakagawa E, Inoue K, Kamada F, Kure S, Goto Y-I, Japanese Mental Retardation Consortium. A loss-of-function mutation in the FTSJ1 gene causes nonsyndromic X-linked mental retardation in a Japanese family. Am J Med Genet B Neuropsychiatr Genet 2008; 147B:479-84; PMID:18081026[http://dx.doi.org/10.1002/ajmg.b.30638]
- Dai L, Xing L, Gong P, Zhang K, Gao X, Zheng Z, Zhou J, Guo Y, Guo S, Zhang F. Positive association of the FTSJ1 gene polymorphisms with nonsyndromic X-linked mental retardation in young Chinese male subjects. J Hum Genet 2008; 53:592-7; PMID:18401546[http://dx.doi.org/10.1007/s10038-008-0287-x]
- Gong P, Li J, Dai L, Zhang K, Zheng Z, Gao X, Zhang F. Genetic Variations in FTSJ1Influence Cognitive Ability in Young Males in the Chinese Han Population. J Neurogenet 2008; 22:277-87; PMID:19012053[http://dx.doi.org/10.1080/01677060802337299]
- Honda S, Hayashi S, Imoto I, Toyama J, Okazawa H, Nakagawa E, Goto Y-I, Inazawa J, Consortium JMR. Copy-number variations on the X chromosome in Japanese patients with mental retardation detected by array-based comparative genomic hybridization analysis. J Hum Genet 2010; 55:590-9; PMID:20613765[http://dx.doi.org/10.1038/jhg.2010.74]
- El-Hattab AW, Bournat J, Eng PA, Wu J, Walker BA, Stankiewicz P, Cheung SW, Brown CW. Microduplication of Xp11.23p11.3 with effects on cognition, behavior, and craniofacial development. Clin Genet 2010; 79:531-8; http://dx.doi.org/10.1111/j.1399-0004.2010.01496.x
- Bonnet C, Grégoire MJ, Brochet K, Raffo E, Leheup B, Jonveaux P. Pure de-novo 5 Mb duplication at Xp11.22-p11.23 in a male: phenotypic and molecular characterization. J Hum Genet 2006; 51:815-21; PMID:16900295
- Abbasi-Moheb L, Mertel S, Gonsior M, Nouri-Vahid L, Kahrizi K, Cirak S, Wieczorek D, Motazacker MM, Esmaeeli-Nieh S, Cremer K, et al. Mutations in NSUN2 cause autosomal- recessive intellectual disability. Am J Hum Genet 2012; 90:847-55; PMID:22541559[http://dx.doi.org/10.1016/j.ajhg.2012.03.021]
- Fahiminiya S, Almuriekhi M, Nawaz Z, Staffa A, Lepage P, Ali R, Hashim L, Schwartzentruber J, Abu Khadija K, Zaineddin S, et al. Whole exome sequencing unravels disease-causing genes in consanguineous families in Qatar. Clin Genet 2014; 86:134-41; PMID:24102521[http://dx.doi.org/10.1111/cge.12280]
- Khan MA, Rafiq MA, Noor A, Hussain S, Flores JV, Rupp V, Vincent AK, Malli R, Ali G, Khan FS, et al. Mutation in NSUN2, which Encodes an RNA Methyltransferase, Causes Autosomal-Recessive Intellectual Disability. Am J Hum Genet 2012; 90:856-63; PMID:22541562[http://dx.doi.org/10.1016/j.ajhg.2012.03.023]
- Martinez FJ, Lee JH, Lee JE, Blanco S, Nickerson E, Gabriel S, Frye M, Al-Gazali L, Gleeson JG. Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J M Gen 2012; 49:380-5; PMID:22577224[http://dx.doi.org/10.1136/jmedgenet-2011-100686]
- Hussain S, Tuorto F, Menon S, Blanco S, Cox C, Flores JV, Watt S, Kudo NR, Lyko F, Frye M. The mouse cytosine-5 RNA methyltransferase NSun2 is a component of the chromatoid body and required for testis differentiation. Mol Cell Biol 2013; 33:1561-70; PMID:23401851[http://dx.doi.org/10.1128/MCB.01523-12]
- Chi L, Delgado-Olguín P. Expression of NOL1/NOP2/sun domain (Nsun) RNA methyltransferase family genes in early mouse embryogenesis. Gene Expr Pattern 2013; 13:319-27; PMID:23816522[http://dx.doi.org/10.1016/j.gep.2013.06.003]
- Hussain S, Sajini AA, Blanco S, Dietmann S, Lombard P, Sugimoto Y, Paramor M, Gleeson JG, Odom DT, Ule J, et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determinesIts processing into regulatory small RNAs. Cell Rep 2013; 4:255-61
- Zhang X, Liu Z, Yi J, Tang H, Xing J, Yu M, Tong T, Shang Y, Gorospe M, Wang W. The tRNA methyltransferase NSun2 stabilizes p16INK⁴ mRNA by methylating the 3'-untranslated region of p16. Nat Commun 2012; 3:712; PMID:22395603[http://dx.doi.org/10.1038/ncomms1692]
- Blanco S, Dietmann S, Flores JV, Hussain S, Kutter C, Humphreys P, Lukk M, Lombard P, Treps L, Popis M, et al. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J 2014; PMID:25063673
- Gillis D, Krishnamohan A, Yaacov B, Shaag A, Jackman JE, Elpeleg O. TRMT10A dysfunction is associated with abnormalities in glucose homeostasis, short stature and microcephaly. J Med Gen 2014; 51:581-6; PMID:25053765[http://dx.doi.org/10.1136/jmedgenet-2014-102282]
- Igoillo-Esteve M, Genin A, Lambert N, Désir J, Pirson I, Abdulkarim B, Simonis N, Drielsma A, Marselli L, Marchetti P, et al. tRNA methyltransferase homolog gene TRMT10A mutation in young onset diabetes and primary microcephaly in humans. PLoS Genet 2013; 9:e1003888; PMID:24204302
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 2014; 10:93-5; PMID:24316715[http://dx.doi.org/10.1038/nchembio.1432]
- Ping X-L, Sun B-F, Wang L, Xiao W, Yang X, Wang W-J, Adhikari S, Shi Y, Lv Y, Chen Y-S, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 2014; 24:177-89; PMID:24407421[http://dx.doi.org/10.1038/cr.2014.3]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 2014; 16:191-8; PMID:24394384[http://dx.doi.org/10.1038/ncb2902]
- Horiuchi K, Umetani M, Minami T, Okayama H, Takada S, Yamamoto M, Aburatani H, Reid PC, Housman DE, Hamakubo T, et al. Wilms' tumor 1-associating protein regulates G2/M transition through stabilization of cyclin A2 mRNA. Proc Natl Acad Sci USA 2006; 103:17278-83.
- Fukusumi Y, Naruse C, Asano M. Wtap is required for differentiation of endoderm and mesoderm in the mouse embryo. Dev Dyn 2008; 237:618-29; PMID:18224709[http://dx.doi.org/10.1002/dvdy.21444]
- Higa-Nakamine S, Suzuki T, Uechi T, Chakraborty A, Nakajima Y, Nakamura M, Hirano N, Suzuki T, Kenmochi N. Loss of ribosomal RNA modification causes developmental defects in zebrafish. Nucleic Acids Res 2011; PMID:21908402
- Alazami AM, Hijazi H, Al-Dosari MS, Shaheen R, Hashem A, Aldahmesh MA, Mohamed JY, Kentab A, Salih MA, Awaji A, et al. Mutation in ADAT3, encoding adenosine deaminase acting on transfer RNA, causes intellectual disability and strabismus. JM Gen 2013; 50:425-30; PMID:23620220[http://dx.doi.org/10.1136/jmedgenet-2012-101378]
- Emmerich B, Zubrod E, Weber H, Maubach PA, Kersten H, Kersten W. Relationship of queuine-lacking transfer RNA to the grade of malignancy in human leukemias and lymphomas. Cancer Res 1985; 45:4308-14; PMID:4028017
- Kretz KA, Katze JR, Trewyn RW. Guanine analog-induced differentiation of human promyelocytic leukemia cells and changes in queuine modification of tRNA. Mol Cell Biol 1987; 7:3613-9; PMID:3479681
- Rodriguez V, Chen Y, Elkahloun A, Dutra A, Pak E, Chandrasekharappa S. Chromosome 8 BAC array comparative genomic hybridization and expression analysis identify amplification and overexpression of TRMT12 in breast cancer. Genes Chromosomes Cancer 2007; 46:694-707; PMID:17440925[http://dx.doi.org/10.1002/gcc.20454]
- Pathak C, Jaiswal YK, Vinayak M. Modulation in the activity of lactate dehydrogenase and level of c-Myc and c-Fos by modified base queuine in cancer. Cancer Biol Ther 2008; 7:85-91; PMID:18347422[http://dx.doi.org/10.4161/cbt.7.1.5133]
- Vachon CM, Sellers TA, Carlson EE, Cunningham JM, Hilker CA, Smalley RL, Schaid DJ, Kelemen LE, Couch FJ, Pankratz VS. Strong evidence of a genetic determinant for mammographic density, a major risk factor for breast cancer. Cancer Res 2007; 67:8412-8; PMID:17804758[http://dx.doi.org/10.1158/0008-5472.CAN-07-1076]
- Shimada K, Nakamura M, Anai S, De Velasco M, Tanaka M, Tsujikawa K, Ouji Y, Konishi N. A novel human AlkB homologue, ALKBH8, contributes to human bladder cancer progression. Cancer Res 2009; 69:3157-64; PMID:19293182[http://dx.doi.org/10.1158/0008-5472.CAN-08-3530]
- Berg M, Agesen TH, Thiis-Evensen E, group IN-S, Merok MA, Teixeira MR, Vatn MH, Nesbakken A, Skotheim RI, Lothe RA. Distinct high resolution genome profiles of early onset and late onset colorectal cancer integrated with gene expression data identify candidate susceptibility loci. Mol Cancer 2010; 9:100; PMID:20459617[http://dx.doi.org/10.1186/1476-4598-9-100]
- Kerr SJ, Borek E. Regulation of the tRNA methyltransferases in normal and neoplastic tissues. Adv Enzyme Regul 1973; 11:63-77; PMID:4799201[http://dx.doi.org/10.1016/0065-2571(73)90009-5]
- Dirheimer G, Baranowski W, Keith G. Variations in tRNA modifications, particularly of their queuine content in higher eukaryotes. Its relation to malignancy grading. Biochimie 1995; 77:99-103; PMID:7599283[http://dx.doi.org/10.1016/0300-9084(96)88111-9]
- Okada N, Shindo-Okada N, Sato S, Itoh YH, Oda K, Nishimura S. Detection of unique tRNA species in tumor tissues by Escherichia coli guanine insertion enzyme. Proc Natl Acad Sci USA 1978; 75:4247-51.
- Morris RC, Galicia MC, Clase KL, Elliott MS. Determination of queuosine modification system deficiencies in cultured human cells. Mol Gen Meta 1999; 68:56-67; PMID:10479483[http://dx.doi.org/10.1006/mgme.1999.2889]
- Costa A, Pais de Barros JP, Keith G, Baranowski W, Desgres J. Determination of queuosine derivatives by reverse-phase liquid chromatography for the hypomodification study of Q-bearing tRNAs from various mammal liver cells. J Chromatogr B Analyt Technol Biomed Life Sci 2004; 801:237-47; PMID:14751792[http://dx.doi.org/10.1016/j.jchromb.2003.11.022]
- Shindo-Okada N, Terada M, Nishimura S. Changes in amount of hypo-modified tRNA having guanine in place of queuine during erythroid differentiation of murine erythroleukemia cells. Eur J Biochem 1981; 115:423-8; PMID:7238512[http://dx.doi.org/10.1111/j.1432-1033.1981.tb05254.x]
- Huang BS, Wu RT, Chien KY. Relationship of the queuine content of transfer ribonucleic acids to histopathological grading and survival in human lung cancer. Cancer Res 1992; 52:4696-700; PMID:1511436
- Katze JR, Basile B, McCloskey JA. Queuine, a modified base incorporated posttranscriptionally into eukaryotic transfer RNA: wide distribution in nature. Science 1982; 216:55-6; PMID:7063869[http://dx.doi.org/10.1126/science.7063869]
- Pathak C, Jaiswal YK, Vinayak M. Queuine promotes antioxidant defence system by activating cellular antioxidant enzyme activities in cancer. Biosci Rep 2008; 28:73-81; PMID:18290765[http://dx.doi.org/10.1042/BSR20070011]
- Shindo-Okada N, Akimoto H, Nomura H, Nishimura S. Recognition of UAG termination codon by mammalian tyrosine tRNA containing 6-thioqueuine in the first position of the anticodon. Proc Japan Acad 1985; 61(B):94-8.
- Frye M, Watt FM. The RNA Methyltransferase Misu (NSun2) Mediates Myc-Induced Proliferation and Is Upregulated in Tumors. Curr Biol 2006; 16:971-81; PMID:16713953[http://dx.doi.org/10.1016/j.cub.2006.04.027]
- Schaefer M, Hagemann S, Hanna K, Lyko F. Azacytidine inhibits RNA methylation at DNMT2 target sites in human cancer cell lines. Cancer Res 2009; 69:8127-32; PMID:19808971[http://dx.doi.org/10.1158/0008-5472.CAN-09-0458]
- Christian T, Gamper H, Hou YM. Conservation of structure and mechanism by Trm5 enzymes. RNA 2013; 19:1192-9; PMID:23887145[http://dx.doi.org/10.1261/rna.039503.113]
- Songe-Møller L, van den Born E, Leihne V, Vågbø CB, Kristoffersen T, Krokan HE, Kirpekar F, Falnes PØ, Klungland A. Mammalian ALKBH8 possesses tRNA methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol Cell Biol 2010; 30:1814-27; PMID:20123966[http://dx.doi.org/10.1128/MCB.01602-09]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ, Vågbø CB, Shi Y, Wang W-L, Song S-H, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 2013; 49:18-29; PMID:23177736
- Leidel S, Pedrioli PGA, Bucher T, Brost R, Costanzo M, Schmidt A, Aebersold R, Boone C, Hofmann K, Peter M. Ubiquitin-related modifier Urm1 acts as a sulphur carrier in thiolation of eukaryotic transfer RNA. Nature 2009; 458:228-32; PMID:19145231[http://dx.doi.org/10.1038/nature07643]
- Bloom-Ackermann Z, Navon S, Gingold H, Towers R, Pilpel Y, Dahan O. A Comprehensive tRNA Deletion Library Unravels the Genetic Architecture of the tRNA Pool. PLoS Genet 2014; 10:e1004084; PMID:24453985
- Hanada T, Weitzer S, Mair B, Bernreuther C, Wainger BJ, Ichida J, Hanada R, Orthofer M, Cronin SJ, Komnenovic V, et al. CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature 2013; 495:474-80; PMID:23474986[http://dx.doi.org/10.1038/nature11923]
- Ishimura R, Nagy G, Dotu I, Zhou H, Yang X-L, Schimmel P, Senju S, Nishimura Y, Chuang JH, Ackerman SL. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 2014; 345:455-9; PMID:25061210[http://dx.doi.org/10.1126/science.1249749]
- Karaca E, Weitzer S, Pehlivan D, Shiraishi H, Gogakos T, Hanada T, Jhangiani SN, Wiszniewski W, Withers M, Campbell IM, et al. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell 2014; 157:636-50; PMID:24766809[http://dx.doi.org/10.1016/j.cell.2014.02.058]
- Schaffer AE, Eggens VRC, Caglayan AO, Reuter MS, Scott E, Coufal NG, Silhavy JL, Xue Y, Kayserili H, Yasuno K, et al. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell 2014; 157:651-63; PMID:24766810[http://dx.doi.org/10.1016/j.cell.2014.03.049]
- Samaan Z, Anand S, Zhang X, Desai D, Rivera M, Pare G, Thabane L, Xie C, Gerstein H, Engert JC, et al. The protective effect of the obesity-associated rs9939609 A variant in fat mass- and obesity-associated gene on depression. Mol Psychiatry 2012; 18:1281-6; PMID:23164817[http://dx.doi.org/10.1038/mp.2012.160]
- Sobczyk-Kopciol A, Broda G, Wojnar M, Kurjata P, Jakubczyk A, Klimkiewicz A, Ploski R. Inverse association of the obesity predisposing FTO rs9939609 genotype with alcohol consumption and risk for alcohol dependence. Addiction 2010; 106:739-48; PMID:21182554[http://dx.doi.org/10.1111/j.1360-0443.2010.03248.x]
- Haverty PM, Fridlyand J, Li L, Getz G, Beroukhim R, Lohr S, Wu TD, Cavet G, Zhang Z, Chant J. High-resolution genomic and expression analyses of copy number alterations in breast tumors. Genes Chromosomes Cancer 2008; 47:530-42; PMID:18335499[http://dx.doi.org/10.1002/gcc.20558]
- Guan M-X, Yan Q, Li X, Bykhovskaya Y, Gallo-Teran J, Hajek P, Umeda N, Zhao H, Garrido G, Mengesha E, et al. Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am J Hum Genet 2006; 79:291-302; PMID:16826519[http://dx.doi.org/10.1086/506389]
- Zeharia A, Shaag A, Pappo O, Mager-Heckel A-M, Saada A, Beinat M, Karicheva O, Mandel H, Ofek N, Segel R, et al. Acute infantile liver failure due to mutations in the TRMU gene. Am J Hum Genet 2009; 85:401-7; PMID: 19732863[http://dx.doi.org/10.1016/j.ajhg.2009.08.004]
- Blanco S, Kurowski A, Nichols J, Watt FM, Benitah SA, Frye M. The RNA-methyltransferase Misu (NSun2) poises epidermal stem cells to differentiate. PLoS Genet 2011; 7:e1002403
- Schaefer M, Pollex T, Hanna K, Tuorto F, Meusburger M, Helm M, Lyko F. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev 2010; 24:1590-5; PMID:20679393[http://dx.doi.org/10.1101/gad.586710]
- Penalva LO, Ruiz MF, Ortega A, Granadino B, Vicente L, Segarra C, Valcárcel J, Sánchez L. The Drosophila fl(2)d gene, required for female-specific splicing of Sxl and tra pre-mRNAs, encodes a novel nuclear protein with a HQ-rich domain. Genetics 2000; 155:129-39; PMID:10790389